稀土金属苄基配合物高活性催化丙烯腈聚合
2023-11-18罗云杰
徐 森,罗云杰
(宁波大学 材料科学与化学工程学院,浙江 宁波 315211)
聚丙烯腈(PAN)纤维是继聚酯和尼龙之后的第三大合成纤维,聚丙烯腈不仅在纺织工业中占有重要的地位,而且是生产碳纤维的重要前体[1-2].随着聚丙烯腈的分子量增加,聚丙烯腈纤维和以其作为前体合成的碳纤维的性能均有提升[3].目前,聚丙烯腈的工业生产以传统自由基聚合法为主[4],采用以DMSO 为溶剂的均相溶液聚合法一步合成.有专利报道以偶氮二异丁腈为引发剂在DMSO 中进行均相溶液聚合,制得了分子量为(9~15)×104g·mol-1,分子量分布(Mw/Mn)在3.0~3.5 之间的聚合物[5].这种方法具有工艺简单、成本低和聚合产物缺陷少等优点,但聚合过程所使用的有机溶剂会促进链的转移,导致难以得到高分子量和分子量分布较小的聚丙烯腈.当采用水相悬浮聚合或水相乳液聚合法时,体系中加入分散剂,使单体分散到水相中聚合.这种方法抑制了链的转移,有利于制得高分子量和分子量分布稳定的聚丙烯腈.Morris 等[6]将过硫酸铵和焦亚硫酸钠作为引发体系,表面活性剂Dowfax 8390 为乳化剂,进行丙烯腈(AN)与甲基丙烯酸甲酯的水相乳液聚合研究,在25 ℃条件下进行24 h 聚合,可以制得分子量达1.71×106g·mol-1的聚合产物,相对分子量分布在1.6~2.0 范围内.但聚合产物中残留的分散剂和乳化剂通常难以除去,对后续制得高品质碳纤维产生影响,且聚合反应所需时间较长.近年来,活性可控自由基聚合在丙烯腈聚合中得到快速发展.Wei 等[7]采用原子转移自由基聚合法,以有机染料曙红Y 为光催化剂,溴乙酸苯酯和三乙胺为引发剂,通过可见光刺激,在室温条件下合成了分子量大于 5.0×104g·mol-1,分子量分布较小(Mw/Mn=1.69)的聚丙烯腈,但聚合反应所需时间为6 h 以上.这种聚合方式虽然具有聚合产物中不含金属离子等优势,但其聚合效率较低.
相比之下,丙烯腈的阴离子配位聚合具有聚合速率快、活性中心单一等优势[8-12],有利于制备高分子量和分子量分布可控的高聚物.因此,许多种类的金属基引发剂被开发用来催化丙烯腈聚合.Shi 等[3]将二环己基酰胺锂作为引发剂,当n(丙烯腈):n(引发剂)=6 080:1 时,在合适的单体浓度和低温(-60 ℃)条件下可制备高分子量(1.23×106g·mol-1)的PAN;Nakano 等[13]将二烷基镁作为引发剂,在130 ℃条件下,通过往体系中添加氢化二异丁基铝可以将PAN 的等规度(即mm (m:meso))提高(mm=0.63),但此时的单体转化率(聚合产物质量与所用单体质量的比值)只有8.3%,且反应所需温度较高.所以阴离子聚合尚存在不足之处,如反应条件苛刻、转化率低等.因此,开发一种反应条件温和、分子量和分子量分布可控且聚合产物易纯化的催化体系,对聚丙烯腈材料的制备具有重要意义.
稀土金属配合物在聚合体系中一般具有反应条件温和、聚合产物无金属离子残留、聚合产物易纯化、分子量和分子量分布可控以及副反应较少等优点.虽然稀土金属配合物在极性单体(如丙烯酸酯类、内酯[14])和非极性单体(如乙烯、苯乙烯等)聚合中显示出独特的反应性能[15-18],但在丙烯腈聚合中催化活性较低[19-21].Hultzsch 等[20]采用茂基稀土金属二烷基配合物为催化剂,甲苯为溶剂,丙烯腈单体与引发剂物质的量之比为200:1,聚合时间为48 h,仅制得分子量为5.0×103g·mol-1的聚丙烯腈,且反应所需温度为-30 ℃.Roitershtein 等[22]以席夫碱稀土金属配合物/二丁基镁/四甲基乙二胺为引发体系,甲苯为溶剂,在0 ℃条件下催化丙烯腈聚合,丙烯腈单体与引发剂物质的量之比为835:1,聚合时间大于1 h,仅制得分子量为4.2×104g·mol-1、相对分子量分布为2.19 的聚丙烯腈.丙烯腈是一种极性较强的乙烯基单体,本课题组[23-25]在研发用于极性乙烯基单体(如2-乙烯基吡啶)定向聚合的稀土金属催化剂的过程中,发现稀土金属苄基配合物在室温条件下对丙烯腈的聚合具有很高的催化活性.本文研究了稀土金属苄基配合物对丙烯腈的催化聚合,考察了辅助配体、溶剂、金属离子半径以及聚合温度对聚合反应的影响.
1 实验材料与方法
1.1 主要仪器与试剂
所有实验操作均在氩气保护下进行,严格排除空气和水汽,使用Schlenk 技术或在充满氩气的手套箱中进行操作.
Ascend 500 型核磁共振波谱仪(德国Bruker 公司),PL-50 凝胶渗透色谱仪(美国安捷伦公司).
甲苯、四氢呋喃(THF)、正己烷、乙二醇二甲醚(DME)、氯苯(分析纯,国药集团化学试剂有限公司);二苯甲酮(99%,Acros Organics);氢化钙(95%,国药集团化学试剂有限公司);丙烯腈(≥99%,质量分数0.003 5%~0.004 5%的单甲醚二酚作为抑制剂,上海阿拉丁生化科技股份有限公司);氘代苯、氘代氯仿、氘代二甲基亚砜(99.5%,美国CIL);DMF (99.8%,水≤0.005 0%,上海麦克林生化科技股份有限公司).
甲苯、四氢呋喃和正己烷: 先用高温马弗炉活化的分子筛浸泡72 h,然后加入光亮的金属钠丝加热回流除水,直至体系内二苯甲酮指示剂呈深紫色,加热蒸出备用.氯苯、乙二醇二甲醚和丙烯腈用氢化钙干燥48 h 后,减压蒸馏蒸出备用.所有氘代试剂均在使用前数天经光亮的钠丝除水处理.DMF 在使用前经超声过滤脱气处理.
1.2 实验方法
1.2.1 稀土金属配合物的合成
根据文献制备稀土金属苄基配合物(图1):(2,5-Me2C4H2NCH2CH2NC6H5)Sc(CH2C6H4NMe2-o)2(1),(2,5-Me2C4H2NCH2CH2NC6H5)La(CH2C6H4NMe2-o)2(2),(2,5-Me2C4H2NSiMe2NC6H4)Sc(CH2C6H4NMe2-o)2(3),(C5Me5)Sc(CH2C6H4NMe2-o)2(4)[24-26],Ln(CH2C6H4NMe2-o)3(Ln=Sc (5),Y (6),La (7),Lu (8))[27-28].

图1 配合物的结构式
1.2.2 聚丙烯腈的分子量及分子量分布的测定
称取一定量的聚合物,用DMF 作为溶剂,配成质量浓度为1 g·L-1的溶液.使用PL 公司生产的PL-50 凝胶渗透色谱仪测量聚合产物的分子量及分子量分布.测试条件: 两根Mixed-B 凝胶色谱柱串联,柱温设置为40 ℃,以DMF 作为流动相,流动相流速为1.0 mL·min-1,采用聚苯乙烯标准样品对分子量进行校正.
1.2.3 聚丙烯腈的微观结构表征
聚合物的微观结构用13C NMR 谱图进行分析,对主链上次甲基中碳原子出峰在化学位移26.5~28.8 处的峰面积归属和积分,推定聚合物的立构规整性[29].
1.2.4 丙烯腈的聚合反应
聚合反应所需的丙烯腈单体和相应的聚合溶剂在使用前需经过重蒸精制.由于这些稀土金属配合物引发的丙烯腈聚合的操作过程是类似的,下面给出了典型的聚合方法.在手套箱中,向装有磁性搅拌棒的20 mL Schlenk 烧瓶中加入所需量的丙烯腈和氯苯.然后用注射器吸取氯苯溶解的引发剂溶液加到上述烧瓶中.将混合物剧烈搅拌至所需时间,在此期间观察到黏度增加.加入乙醇淬灭反应混合物,然后倒入大量乙醇中沉淀聚合物,在真空下干燥并称重.
2 结果与讨论
图1 所示的稀土金属配合物1~3 在室温下对2-乙烯基吡啶聚合具有高活性[24-25].这些结果表明,稀土金属苄基配合物可能在极性乙烯基的单体聚合反应中有潜在的应用价值.丙烯腈是一种常见的极性乙烯基单体,聚丙烯腈是一种有很好用途的聚合物材料.然而,稀土金属配合物作为催化剂用于丙烯腈聚合的成功例子非常有限[20-21,30-31].
稀土金属苄基配合物对丙烯腈的聚合活性结果见表1.首先考察配合物1~3,中性芳胺基稀土双(苄基)配合物1~3 在25 ℃、氯苯溶剂中引发丙烯腈聚合,得到的聚丙烯腈分子量大于103g·mol-1.尽管配合物1 和2 具有相同的辅助配体,但它们表现出显著不同的聚合活性.例如,使用配合物2 时,当n(丙烯腈):n(Ln)=200:1 时(表1 中的条目2),在几秒内丙烯腈几乎完全转化为聚丙烯腈.相比之下,使用配合物1 作为引发剂(表1 中的条目1)需要1 h 转化率才达到90%.在配合物3 中,辅助配体骨架上芳胺和吡咯之间的连接变短,使其显示出比配合物1 高得多的聚合活性.在相同的聚合条件下,半夹心稀土金属双(苄基)配合物(图1 中配合物4)对丙烯腈聚合也具有高活性.初步聚合结果表明,稀土金属苄基配合物可以高活性催化丙烯腈聚合.然而,辅助配体对聚合活性并没有太大影响(表1 中的条目1、3 和4).因此,使用简单易得的稀土三(苄基)配合物Ln(CH2C6H4NMe2-o)3(Ln=Sc(5),Y(6),La(7),Lu(8))作为丙烯腈聚合的引发剂.结果发现,在室温下这些稀土三苄基配合物均对丙烯腈聚合表现出极高的催化活性,当n(丙烯腈):n(Ln)=200:1 时,聚合反应能够在几秒钟内完成(表1 中的条目5~8).
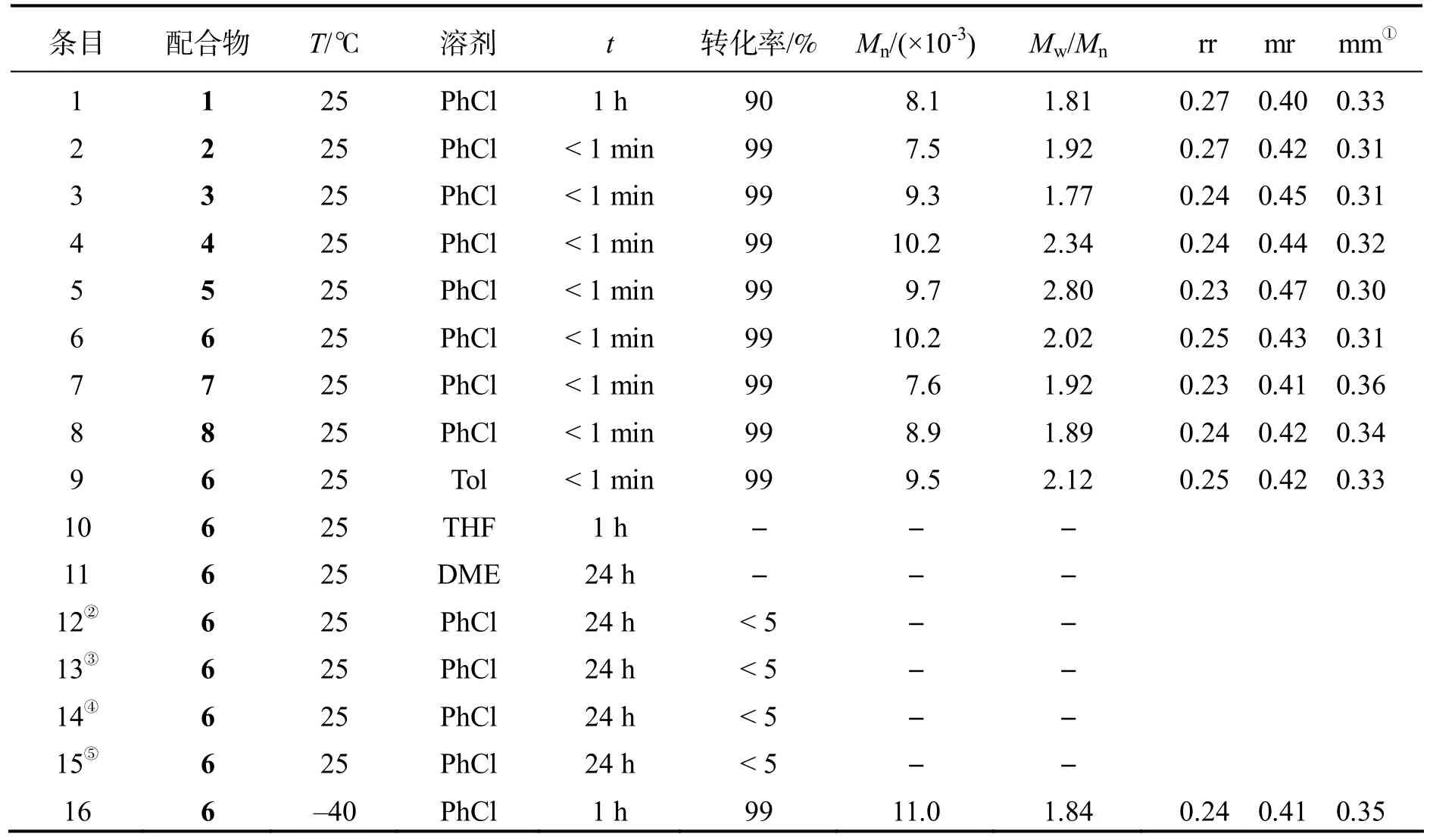
表1 稀土金属苄基配合物催化丙烯腈聚合情况
以上结果说明金属离子半径对聚合活性几乎无影响.因此,使用相对廉价的Y(CH2C6H4NMe2-o)3进一步研究丙烯腈的聚合.结果发现,溶剂对丙烯腈的聚合影响较大.在氯苯和甲苯中,聚合活性明显高于在THF和DME 中进行的聚合反应,DME 作为聚合溶剂,聚合反应很难进行,即使将聚合时间延长至24 h,也得不到聚合物(表1 中的条目9~11).这可能归因于溶剂和丙烯腈对活性中心的配位竞争,THF 和DME 对稀土中心金属的强配位能力阻碍了丙烯腈与活性中心的配位,导致链引发无法进行.值得注意的是,当Y(CH2C6H4NMe2-o)3被硼烷(B(C6F5)3)或硼酸盐([Ph3C][B(C6F5)4])活化,所生成的阳离子配合物无法引发丙烯腈的聚合,这表明金属中心的路易斯酸性增加,加剧了其与单体N原子的相互作用,不利于链的引发和增长(表1 中的条目12~15).对温度对聚合活性的影响进行探究发现,与预期一样,聚合活性随着聚合温度的降低而降低(表1 中的条目16).
对部分所得聚丙烯腈的微观结构进行核磁表征,13C NMR 谱如图2 所示,所得到的聚丙烯腈的微观结构是无规的,辅助配体(条目4、条目5)、聚合溶剂(条目6、条目9)、金属离子半径(条目5、条目6)以及聚合温度对聚合物的规整性影响较小.
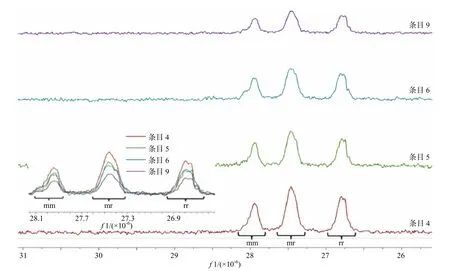
图2 聚丙烯腈13C NMR 谱
3 结论
由于丙烯腈单体中有强吸电子的-CN 基团,要实现聚合过程中的高聚合活性、聚合物的高分子量和高立构选择性仍具有很大挑战性.稀土催化剂对于丙烯腈的聚合通常表现出聚合活性低,聚合反应的选择性差.本文研究发现稀土苄基配合物2、3、4 和Ln(CH2C6H4NMe2-o)3(Ln=Sc (5),Y (6),La (7),Lu (8))能够在室温下高活性催化丙烯腈聚合.虽然所得的聚合物在分子量和立构规整性方面还有待提高,但是,此研究工作可为高效丙烯腈聚合稀土催化剂的研发提供理论和实验依据.
