利用55K芯片进行小麦生育期相关性状的QTL定位及效应分析
2023-11-15温明星王宗宽王海燕王秀娥
温明星 肖 进 徐 涛 孙 丽 王宗宽 王海燕 王秀娥
利用55K芯片进行小麦生育期相关性状的QTL定位及效应分析
温明星1,2,*肖 进2徐 涛2孙 丽2王宗宽2王海燕2王秀娥2
1镇江市农业科学院, 江苏句容 212400;2作物遗传与种质创新利用全国重点实验室 / 南京农业大学, 江苏南京 210095
生育期是决定小麦品种适应性的重要农艺性状, 探索其遗传机制及效应对于小麦品种的选育和推广具有重要意义。本研究以扬麦158和密穗小麦杂交构建的240个重组自交家系(recombinant inbred line, RIL)为材料, 于2~4个环境下对该群体主要生育期性状进行鉴定。利用已构建的高密度遗传图谱, 共检测到52个生育期相关QTL, 分布在2A、2D、3D、4B、4D、5A、5B、5D、6A、6B、7A、7B和7D染色体上。、和均在多年被重复检测到, 分别解释4.56%~46.86%、1.32%~33.40%和2.37%~13.27%的表型变异。-、-、、、、、、、、、和是新发现的位点。效应分析表明, 生育期相关QTL的聚合能不同程度地缩短生育期, 可应用于培育早熟高产小麦品种。
小麦; 生育期; QTL; SNP标记
小麦是世界上最重要的粮食作物之一, 其拔节期、孕穗期、抽穗期、开花期和成熟期等主要生育期与适应性、产量等密切相关, 严重影响小麦种植区域规划、引种及栽培管理措施的选择[1], 一直受到育种工作者的重视。大量研究表明, 小麦适应性主要由春化、光周期和早熟性基因控制[2-4]。其中, 春化基因、、均位于第5同源群染色体长臂[5-7]; 光周期基因、、位于第二同源群染色体上[8-9]; 早熟性基因()独立于春化和光周期基因作用之外, 主要定位于2B、3A、4A、6B、6D、7B等染色体上[10-12]。
生育期是由多基因控制的数量性状, 受环境影响较大, 挖掘与生育期性状相关联的基因位点是分子标记辅助育种及解释基因效应的重要基础。近年来, 随着新的测序技术不断涌现、测序成本的降低以及小麦参考基因组信息的释放, 使得小麦SNP芯片的开发和应用更为普及, 国内外挖掘了大量控制生育期性状的遗传位点。宋彦霞等[13]检测到9个位于2D、3B、3D等染色体上的QTL位点, 对抽穗期贡献率最高达22.91%。茹京娜等[14]利用DH群体在1B、1D、4D、6B、7B和7D染色体上检测到6个抽穗期相关QTL, 对表型贡献率在1.57%~6.72%之间。王克森等[15]以RIL为材料共发现9个抽穗期、7个开花期相关QTL, 解释3.48%~16.93%的表型变异。Chen等[16]利用90K芯片对RIL群体进行基因分型, 在2A、5B、6B、7A和7D染色体上检测到5个稳定的抽穗期QTL, 其中可解释29.35%~41.96%的遗传变异。
随着全球气候变暖及种植业结构调整, 长江中下游麦区稻茬麦播期大幅度推迟, 迫切需要耐迟播早熟小麦品种[17], 解决开花后期高温逼熟而导致产量降低的问题。密穗小麦(Club wheat,Host., 2= 6= 42, 基因组AABBDD)是小麦属一个古老的六倍体裸粒栽培种, 是普通小麦的一级基因库遗传资源[18]。密穗小麦Hiller具有独特的穗部性状, 穗短而宽, 小穗排列极紧密, 生育期偏迟。扬麦158是聚合阿夫、南大2419、胜利麦等多个骨干亲本优良基因而育成的集优质、高产、多抗、广适性于一身的小麦品种, 是长江中下游麦区第6次品种更新换代的主体品种, 也是小麦育种的骨干亲本[19]。扬麦158穗型较大, 生育期较短。
先前针对扬麦158的研究主要集中于产量等农艺性状[20-21]、品质[22]和赤霉病抗性[23]等方面, 其携带的生育期相关基因仍不明确。因此, 本研究利用扬麦158和Hiller衍生的RIL群体构建基于55K SNP芯片的高密度遗传连锁图谱, 结合多环境下的生育期性状, 挖掘与其相关的QTL, 以明确其生育期遗传机制, 为小麦生育期性状基因克隆和分子育种提供有益信息。
1 材料与方法
1.1 试验材料
以扬麦158 (Y158)为母本、密穗小麦Hiller (HL)为父本杂交得到F1, 再利用单粒传法连续自交获得240个家系的F2:10重组自交系(RIL)。扬麦158为中国长江中下游麦区主栽品种和骨干亲本, 全生育期较短; 密穗小麦为古老的六倍体裸粒栽培种, 全生育期较长, 两者在主要生育期性状上具有明显差异。
1.2 田间试验
供试材料于2017—2018年度(E1)、2018—2019年度(E2)、2019—2020年度(E3)和2020—2021年度(E4)种植在江苏丘陵地区镇江农业科学研究所农业科技创新中心。田间种植采用完全随机区组设计, 2次重复, 2行区种植, 行长1.2 m, 行距0.25 m, 生长季内均未发生严重的病虫害及倒伏情况, 田间管理同一般大田生产。
1.3 主要生育期性状测定及数据处理
根据亲本及各家系生育进程, 分别于拔节期(群体50%植株基部第一节间伸长至离地面2 cm, JS)、孕穗期(群体50%旗叶叶片全部抽出叶鞘, 旗叶叶鞘包着的幼穗明显膨大, BS)、抽穗期(群体50%主茎穗的1/3抽出旗叶, HS)、开花期(群体50%植株出现第1朵花, FS)和成熟期(籽粒腊熟, MS)进行性状调查, 以上均为播种日期至相应生育期之间的天数。
利用SPSS 20软件和IciMapping V4.1的ANOVA功能对表型性状进行描述性统计、方差和相关分析。广义遗传率计算公式为:B2=g2/(g2+ge2/+ε2/),g2、ge2和ε2分别表示基因型、基因与环境互作及误差方差,和分别表示环境个数和每个环境的重复数。利用R语言包“lmer”计算各性状的BLUP (Best Linear Unbiased Predictor)值, 计算模型为= lmer (trait~(1|LINE) + (1|LOC) + (1|YEAR) + (1| LOC:YEAR) + (1|LINE:LOC) + (1|LINE:YEAR)), 其中trait代表性状值, LOC代表地点, YEAR代表年份, REP代表重复次数。
1.4 遗传图谱构建及QTL定位
试验使用Wen等[24]基于55K SNP芯片构建的高密度遗传连锁图谱。利用QTL IciMapping V4.1中的完备区间作图法(inclusive composite interval mapping, ICIM-ADD), 扫描步长(step)为1 cM, 逐步回归标记进入的概率(PIN)为0.001, LOD阈值设为2.5, 结合各年度表型及BLUP值, 依据连锁图谱进行目标性状的QTL定位。采用中国春参考基因组2.1[25]查询前人报道的QTL物理位置, 并与本试验所定位到的QTL进行比较。QTL命名方法遵循国际常用命名方法(https://wheat.pw.usda.gov/ggpages/wgc/98/Intro.htm)。
2 结果与分析
2.1 生育期表型变异
在2~4个检测环境中及BLUP值, 母本扬麦158在拔节期、孕穗期、抽穗期、开花期和成熟期上均早于父本密穗小麦。5个性状在RIL群体中存在广泛的遗传变异, 其变异系数BLUP值均低于4% (表1)。遗传率分析结果表明, 拔节期和孕穗期的遗传率较高, 大于90%; 开花期遗传率次之, 为82%; 抽穗期和成熟期稍低, 均小于80%。RIL家系各生育期BLUP值在各环境间相关系数均达极显著正相关(表2), 表型相对稳定。
利用最优线性无偏估计BLUP对多年表型数据进行拟合, 去除环境因素对表型的影响, 对5个目标性状表型数据频次统计分析发现, 所有性状表型的BLUP值均呈现正态分布(图1)。
2.2 生育期性状QTL定位
利用2~4个环境及其BLUP值的表型数据, 共定位到52个主要生育期性状相关QTL, 分布在13条染色体上, 控制拔节期(9个)、孕穗期(12个)、抽穗期(10个)、开花期(12个)和成熟期(9个), 其中3个位点对表型变异的解释率(PVE)大于10%, 为主效QTL (表3和附表1)。

表1 主要生育期性状表型分析
E1、E2、E3、E4和BLUP分别表示2017–2018年度、2018–2019年度、2019–2020年度、2020–2021年度和最优线性无偏估计值。Y158: 扬麦158; HL: Hiller。CV: 变异系数。
E1, E2, E3, E4, and BLUP refer to the environments of 2017–2018, 2018–2019, 2019–2020, 2020–2021, and the best linear unbiased estimations, respectively. Y158: Yangmai 158; HL: Hiller. CV: coefficient of variation.

表2 主要生育期性状相关性分析
**:< 0.01.

图1 主要生育期性状表型分析
数据为各性状在2~4个环境下的最优线性无偏估计值。
The best linear unbiased estimations across 2–4 environments for each trait was used.
9个控制拔节期的QTL分布在2A、2D、4D、5B、5D、6A和7B染色体上, 其中2个贡献率大于10%。、、、和在2个及以上环境中被检测到, 其LOD值分别为6.08、4.02、17.71、42.72和4.69, PVE值分别为4.01%、1.96%、12.62%、36.85%和2.69%, 加性效应为负, 优异等位基因来自扬麦158。
12个控制孕穗期的QTL位于2A、2D、5A、5D、6A、6B、7A、7B和7D染色体上,、、和是最为稳定的QTL, 可在5个环境中被检测到, 单个位点对表型的解释率为1.65%~26.52%, 其加性效应均来自扬麦158;可在4个环境中被检测到, 解释3.10%-6.58%的表型变异, 加性效应为正, 优异等位基因来自密穗小麦。
10个控制抽穗期的QTL分布在2A、2D、5B、5D、6A、7A、7B和7D染色体上。其中6个QTL的加性效应为负, 优异等位基因来自扬麦158; 4个QTL的加性效应为正, 优异等位基因来自密穗小麦。、、、和可在所有环境中被检测到, 解释3.14%~33.40%的表型变异;在3个环境中被检测到, 解释2.52%~2.77%的表型变异;在2个环境中被检测到, 解释1.86%~2.83%的表型变异; 其余3个QTL仅在单环境中被检测到, 解释2.03%~2.33%的表型变异。
12个控制开花期的QTL分布在2A、2D、5A、5B、5D、7A、7B和7D染色体上, 其中10个QTL的加性效应为正, 其优异等位基因来自密穗小麦;
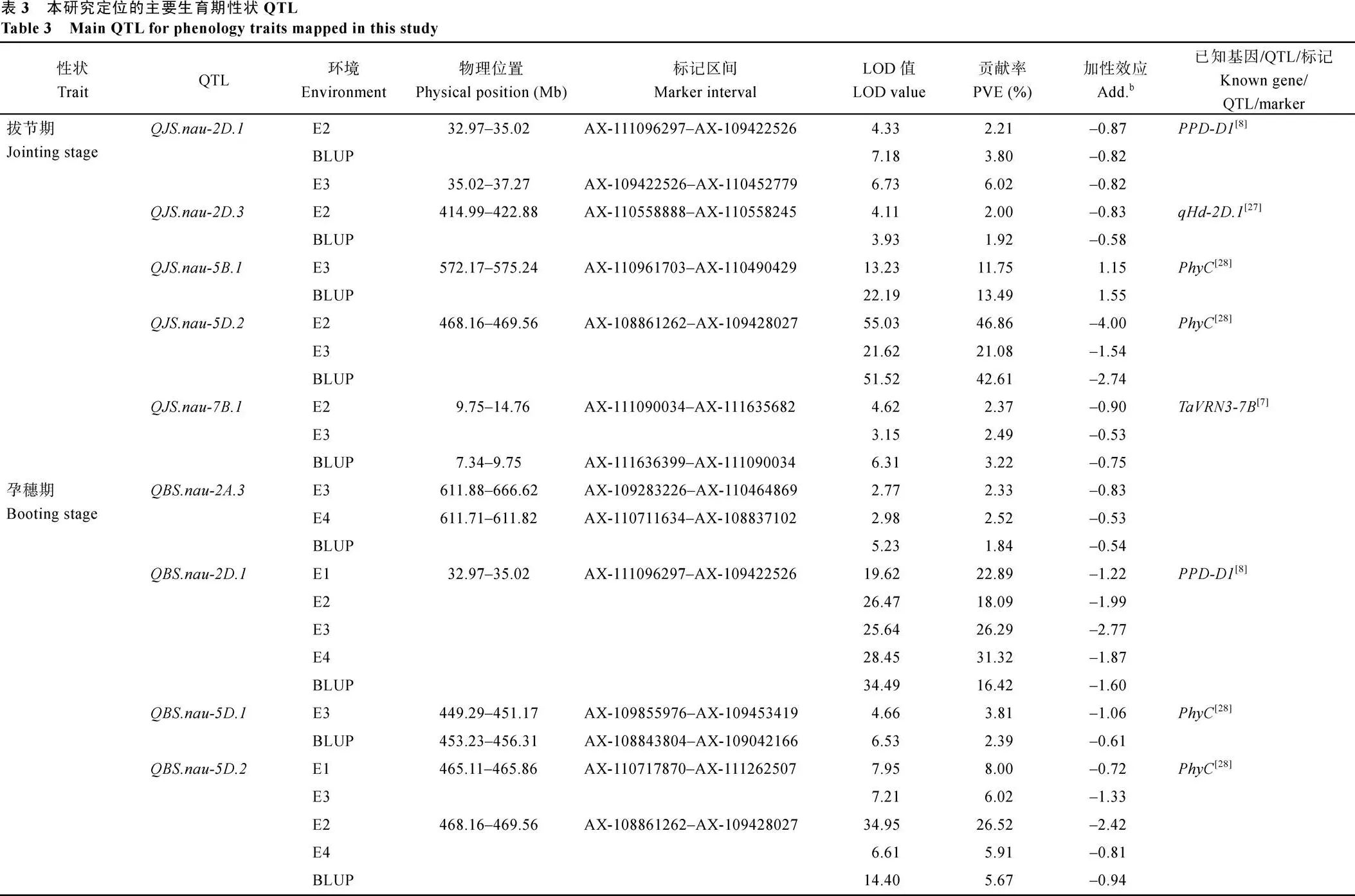
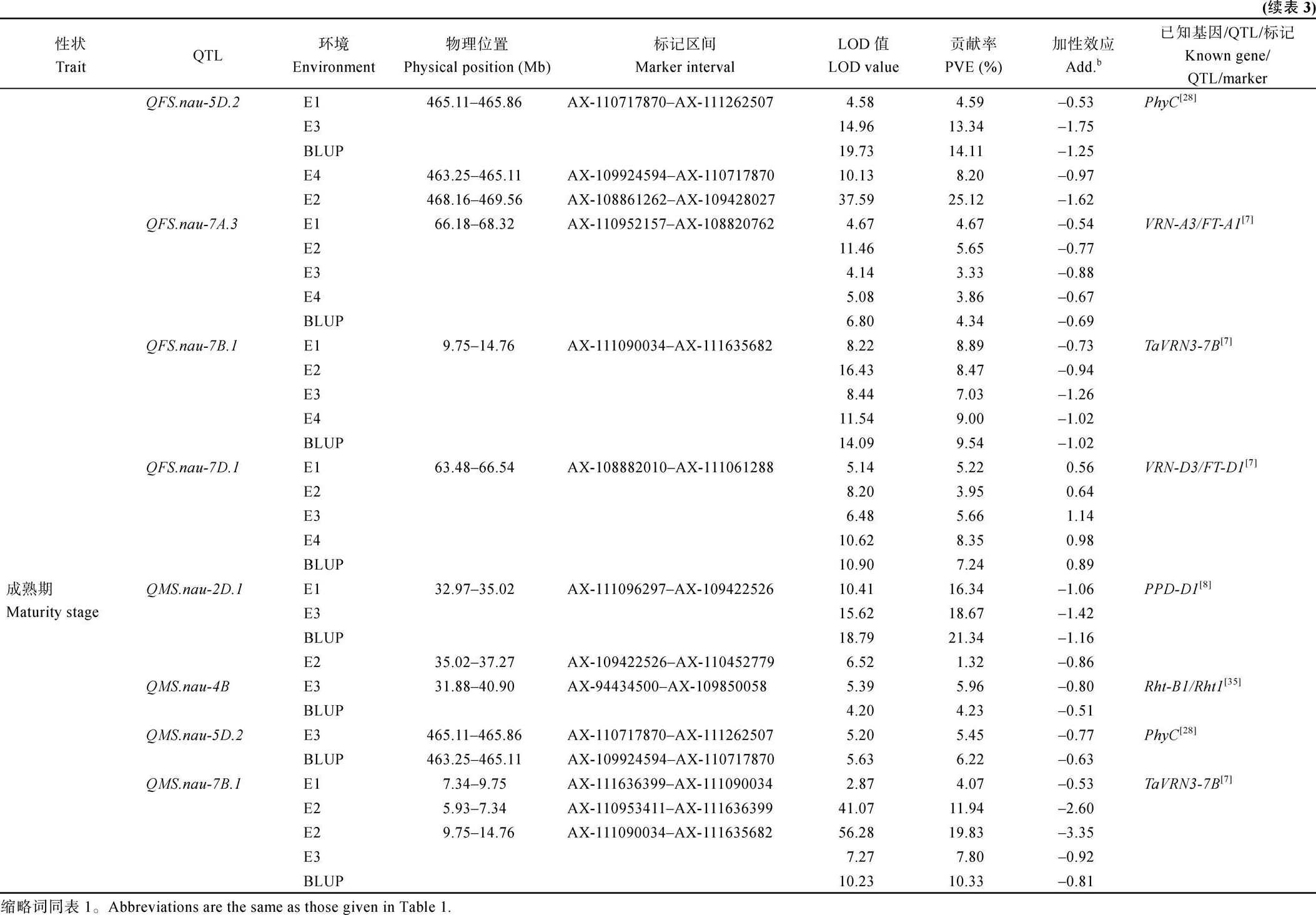
2个QTL的加性效应为负, 优异等位基因来自扬麦158。、、、和可在所有环境中被检测到, 分别解释16.10%~32.00%、4.59%~ 25.12%、3.33%~5.65%、7.03%~9.54%和3.95%~ 8.35%的表型变异;和可在3个环境中被检测到, 解释2.11%~4.53%的表型变异;和可在2个环境中被检测到, 解释2.01%~3.90%的表型变异; 其余3个QTL仅在单环境中被检测到, 解释1.47%~2.40%的表型变异。
9个控制成熟期的QTL分布在2D、3D、4B、4D、5D、6B、7B和7D染色体上, 其中7个QTL的加性效应为负, 优异等位基因来自扬麦158; 2个QTL的加性效应为正, 优异等位基因来自密穗小麦。和可在4个环境中检测到, 分别解释1.32%~21.34%、4.07%~19.83%的表型变异;和可在2个环境中扫描到, 分别解释4.23%~5.96%、5.45%~ 6.22%的表型变异; 其他5个QTL仅在单环境中被检测到, 解释0.71%~3.76%的表型变异。
2.3 RIL群体(扬麦158/密穗小麦)中定位的QTL簇
比较检测到的52个QTL的定位信息, 发现分布在2A、2D、5B、5D、6A、7A、7B和7D上10个QTL同时调控2个及以上的生育期性状(表4), 推测它们为紧密连锁或一因多效的QTL簇。
本研究在2D、5D和7B上定位到涉及所有研究性状的3个QTL簇, 分别位于32.97~35.02 Mb、463.25~469.56 Mb和7.34~14.76 Mb的物理区间, LOD平均值分别为22.31、19.06和14.41, 平均解释20.16%、15.25%和9.02%的表型变异(图2)。在其他染色体上定位到7个QTL簇。其中, 位于7D染色体63.48~66.64 Mb物理区间的QTL簇同时与孕穗期、抽穗期、开花期和成熟期相关, 平均LOD值6.89,平均解释5.07%的表型变异; 位于7A染色体短臂66.18~68.32 Mb物理区间的QTL簇同时调控孕穗期、抽穗期和开花期, 平均LOD值5.25, 平均解释3.61%的表型变异。5个QTL簇与2个性状相关, 2A染色体611.71~611.82 Mb物理区间的QTL簇同时调控孕穗期和抽穗期, 平均LOD值3.60、平均解释2.28%的表型变异; 2D长臂414.98~422.88 Mb物理区间的QTL簇同时调控拔节期和开花期, 平均LOD值4.15%、平均解释2.31%的表型变异; 5B染色体593.48~595.05 Mb区间的QTL簇同时调控抽穗期和开花期, 平均LOD值4.33、平均解释3.24%的表型变异; 5D染色体长臂449.29~456.31 Mb物理区间的QTL簇同时调控孕穗期和成熟期, 平均LOD值4.93、平均解释2.30%的表型变异; 6A染色体短臂6.27~12.23 Mb物理区间的QTL簇同时调控孕穗期和抽穗期, 平均LOD值2.77、平均解释2.55%的表型变异。

表4 同时调控2个及以上性状的QTL簇

图2 2D、5D和7B染色体上定位的生育期性状QTL
2.4 不同QTL组合的遗传效应分析
选择在4个及以上环境中可稳定检测到的QTL以及贡献率较大(>10%)的QTL位点, 分析不同QTL组合对目标性状表型的遗传效应(图3)。
拔节期QTL平均贡献率高达36.85%, 作为主效的QTL, 根据其基因分型结果, 可将240个家系分为2种类型。分析不同类型间拔节期BLUP值的差异, 结果表明, 携带该QTL优异等位基因的家系共118个, 比未携带该等位基因的家系拔节期显著提前4.89%。
孕穗期QTL和可在所有环境中被检测到, 是稳定的QTL, 根据其基因分型结果, 将240个家系分为4种类型。分析不同类型间孕穗期BLUP值的差异, 发现同时携带两个QTL优异等位基因的家系45个、单独携带或优异等位基因的家系分别为70个、63个, 均未携带上述两QTL的家系为52个。同时聚合2个QTL优异等位基因较单独携带和优异等位基因的家系孕穗期分别显著提前1.32%、2.28%, 较未携带任一QTL优异等位基因家系提前3.65%; 单独携带任一QTL优异等位基因家系比未携带任一QTL优异等位基因家系分别显著提前2.30%、1.34%; 两个QTL之间孕穗期无显著差异。
抽穗期QTL和在5个环境中被检测到, 根据其基因分型结果, 将240个家系分为4种类型。分析不同类型间抽穗期BLUP值的差异, 结果表明, 同时携带两个QTL优异等位基因的家系为51个、单独携带或优异等位基因的家系分别为72个、66个, 均未携带上述两个QTL的家系46个。同时聚合两个QTL优异等位基因较单独携带或家系抽穗期分别显著提前2.68%、3.21%, 比未携带任一QTL优异等位基因家系显著提前5.16%; 单独携带任一QTL优异等位基因比无优异等位基因家系抽穗期分别显著提前2.42%、1.89%; 2个QTL之间的抽穗期无明显差异。
开花期QTL和在5个环境中均被检测到, 根据其基因分型结果, 240个家系共分为4种类型。分析不同类型间开花期BLUP值的差异, 表明同时携带两个QTL优异等位基因的家系共58个、单独携带任一QTL优异等位基因家系分别为64个、61个, 均未携带以上2个QTL优异等位基因家系的个数为52。同时聚合2个QTL优异等位基因较单独携带或使得开花期分别显著提前2.31%、2.72%, 比未携带目标优异等位基因家系提前4.35%;单独携带或比未携带目标优异等位基因家系的开花期分别明显提前1.99%、1.58%; 单独携带任一目标优异等位基因家系之间的开花期无明显变化。
成熟期QTL和在4个环境中被检测到, 其基因分型将240个家系分为4种类型。研究不同类型间成熟期BLUP值之间的差异, 发现同时携带两个QTL优异等位基因的家系为51个, 单独携带任一QTL优异等位基因的家系分别为67个、71个, 未携带目标优异等位基因的家系为50个。同时聚合两个QTL优异等位基因的家系较单独携带或在成熟期上分别显著提前1.24%、1.58%, 较未携带任一QTL优异等位基因家系显著提前2.43%; 单独携带或优异等位基因家系的成熟期较未携带目标优异等位基因家系分别显著提前1.42%、1.03%; 携带或优异等位基因之间的成熟期无显著差异。
3 讨论
3.1 生育期QTL的比较分析
本研究对5个生育期性状进行QTL定位与分析, 共检测到52个相关位点, 分布在2A、2D、3D、4B、4D、5A、5B、5D、6A、6B、7A、7B和7D染色体上, 单个QTL可解释1.32%~46.86%的表型变异。通过对以上主要QTL位点与前人已报道位点的异同进行比较, 可为解析不同位点遗传效应及分子标记辅助选择提供可靠依据。
3.1.1 QTL定位区间内已报道的功能基因分析
根据QTL定位区间内基因的注释信息(Chinese Spring RefSeq v1.0, http://www.wheatgenome.org/; IWGSC, 2018), 比较定位区间内已报道相关基因的物理位置发现, 10个已报道的重要功能基因位于本研究的QTL定位区间(表3和附表1)。包括: 淀粉合成相关基因[26](); 光周期基因[8](); 抽穗和开花基因[28](、、); 春化基因[7]()、[7]()、[7]()和[33](); 穗密度调控基因[24](); “绿色革命”基因[35](); 抽穗期相关基因[29]()。这一方面验证了本研究QTL分析的结果, 也表明这些基因在不同群体或背景中可能具有保守的功能。
3.1.2 本研究发掘的QTL与已知生育期QTL比较
: 本位点可在两个以上环境中被检测到, 同时与孕穗期和抽穗期相关, 是来源于扬麦158的稳定QTL。Wang等[36]利用和尚麦和豫8679构建的RIL群体在2A染色体上定位到成熟期相关位点-, 侧翼SSR标记为Xbarc124, 根据比对结果, 其物理位置大概为316.87 Mb, 与相距甚远, 推测可能为新的QTL位点。
: 该位点同时与抽穗期和开花期关联, 均在3个环境下被检测到, 位于SNP标记AX-108736770与AX-109321892之间, 物理区间为593.48~595.05 Mb。Thambugala等[37]报道5BL上存在影响抽穗期的位点, 处于90K SNP标记BS00011514_51–IACX5702之间, 两者相距物理位置20 Mb, 可视为新发现的稳定生育期位点。
: 本研究在2A长臂上检测到1个两环境下稳定存在的开花期相关微效位点, 其物理位置为557.00~568.31 Mb。Chen等[16]使用Yi5029和农大4332构建的RIL群体在2A短臂上定位了抽穗期相关位点, 物理位置为199.58 Mb, 二者应为不同位点。
其他均为仅在单环境中扫描到的QTL, 经比对也可能为本文所新发现的位点。如其BLUP值条件下对孕穗期的贡献率高达29.47%, 与Addisson等[38]、Fatima等[39]分别报道的-、-位点物理区间均无法重叠, 可能属于不同位点;仅在E3环境下存在, 对拔节期表型的解释率为5.69%, 在距离本位点20 Mb左右的物理位置存在着控制根长和株高相关的基因[40], 由此可见, 两者并不相同。其余QTL、、、、、和等7个位点均只具有微效效应, 其贡献率均小于4%, 经比较分析[41], 尚未发现有相同或近似的相关报道, 同样可视为新发现的位点。
此外, 本研究检测到的有些QTL位点是前人已报道的。为在两个以上环境中被检测的稳定QTL, 同时与拔节期、开花期有关, 定位于414.99~422.88 Mb之间; 同样在该区段, Fan等[27]定位到了抽穗期相关QTL, 两者应为相同位点。、、、和分别与生育期相关QTL或标记[30]、[31]、[32]、[32]以及[34]物理位置相同或相近, 可能为一致的微效位点。
3.2 遗传改良与QTL/基因聚合
本研究利用遗传贡献率较高或稳定表达的QTL进行遗传效应分析, RIL家系中携带有1个QTL等位基因比无QTL等位基因、携带2个QTL等位基因比携带1个QTL或无QTL等位基因均使得拔节期、孕穗期、抽穗期、开花期和成熟期不同程度显著提前, 表明多个QTL/基因聚合可显著改良生育期性状。在这方面, 其他性状上具有诸多类似研究。胡文静等[42]认为相较于单个位点, 多个抗性QTL的聚合可显著降低赤霉病严重度。Chen等[43]发现7DS上的QTL ()控制千粒重和籽粒宽度, 在剩余杂合体中可分别提高千粒重12.79%~ 21.75%、粒宽4.10%~8.47%。Lin等[44]通过H461/中国春构建的重组自交系群体在2DL上定位到穗粒数主效QTL, 并将其转化为KASP标记, 在3个不同遗传背景群体中进行效应验证, 发现携带该QTL位点的材料比未携带该位点材料能平均增加17.01%的穗粒数。同时, 将历年RIL群体可育小穗数和千粒重BLUP值[24]进行方差分析, 显示是否携带成熟期QTL群体家系之间的产量性状无显著性差异(<0.05)。因此, 将多个生育期性状QTL/基因进行聚合, 对于培育早熟高产小麦品种的分子标记辅助选择育种格外具有意义。
4 结论
利用扬麦158/密穗小麦RIL群体, 在多个环境下对生育期进行鉴定, 结合55K SNP芯片共检测到52个生育期相关位点, 其中12个可能为新的位点、3个具有较大的表型贡献率, QTL聚合可不同程度地缩短生育期。
[1] 王一钊, 刘玉秀, 孟天琪, 魏仕, 张正茂. 小麦温光发育及相关基因研究进展. 麦类作物学报, 2023, 43: 14–25. Wang Y Z, Liu Y X, Meng T Q, Wei S, Zhang Z M. Research progress on thermo-photoperiod development and related genes in wheat., 2023, 43: 14–25 (in Chinese with English abstract).
[2] Worland A J. The influence of flowering time genes on environmental adaptability in European wheats., 1996, 89: 49–57.
[3] Worland A J, Borner A, Korzun V, Li W M, Petrovic S, Sayers E J. The influence of photoperiod genes on the adaptability of European winter wheats., 1998, 100: 385–394.
[4] Snape J W, Butterworth K, Whitechurch E, Worland A J. Waiting for fine times: genetics of flowering time in wheat., 2001, 119: 185–190.
[5] Yan L L, Loukoianov A, Tranquilli G, Helguera M, Fahima T, Dubcivsky J. Positional cloning of the wheat vernalization gene., 2003, 100: 6263–6268.
[6] Yan L L, Loukoianov A, Blechl A, Tranquilli G, Ramakrishna W, SanMiguel P, Bennetzen J L, Echenique V, Dubcovsky J. The wheatgene is a flowering repressor down-regulated by vernalization., 2004, 303: 1640–1644.
[7] Yan L L, Fu D, Li C, Blechl A, Tranquilli G, Bonafede M, Sanchez A, Valarik M, Yasuda S, Dubcovsky J. The wheat and barley vernalization geneis an orthologue of FT., 2006, 103: 19581–19586.
[8] Beales J, Turner A, Griffiths S, Snape J W, Laurie D A. Aresponse regulator is mis expressed in the photoperiod insensitivemutant of wheat (L.)., 2007, 115: 721–733.
[9] Wilhelm E P, Turner A S, Laurie D A. Photoperiod insensitivemutation in tetraploid wheat (L.)., 2009, 118: 285–294.
[10] Hoodgendoorn J. A reciprocal F1monosomic analysis of the genetic control of time of ear emergence, number of leaves and number of spikelets in wheat (L.).1985, 34: 545–558.
[11] Scarth R, Law C N. The location of the photoperiod gene,and an additional genetic factor for ear emergence time on chromosome 2B of wheat., 1983, 51: 607–619.
[12] Zemetra R S, Morris R, Schmidt J W. Gene location for heading date using reciprocal chromosome substitutions in winter wheat., 1986, 26: 531–533.
[13] 宋彦霞, 景蕊莲, 霍纳新, 任正隆, 贾继增. 普通小麦(L.)不同作图群体抽穗期QTL分析. 中国农业科学, 2006, 39: 2186–2193. Song Y X, Jing R L, Huo N X, Ren Z L, Jia J Z. Detection of QTLs for heading in common wheat (L.) using different population., 2006, 39: 2186–2193 (in Chinese with English abstract).
[14] 茹京娜, 于洋, 董凡凡, 吕欣迪, 曹译文, 纪志芳, 陈梦楠, 史雨刚, 王曙光, 孙黛珍. 小麦抽穗期QTL及其与环境的互作. 麦类作物学报, 2014, 34: 1185–1190. Ru J N, Yu Y, Dong F F, Lyu X D, Cao Y W, Ji Z F, Chen M N, Shi Y G, Wang S G, Sun D Z. Analysis of QTL for heading date and interaction effects with environments in wheat., 2014, 34: 1185–1190 (in Chinese with English abstract).
[15] 王克森, 董爽爽, 李法计, 郭军, 台述强, 王利彬, 程敦公, 穆平, 刘建军, 李豪圣, 赵振东, 曹新有, 张玉梅. 小麦抽穗和开花期相关QTL定位与分析. 山东农业科学, 2020, 52(1): 17–23. Wang K S, Dong S S, Li F J, Guo J, Tai S Q, Wang L B, Cheng D G, Mu P, Liu J J, Li H S, Zhao Z D, Cao X Y, Zhang Y M. QTL mapping and analysis of heading time and flowering time of wheat., 2020, 52(1): 17–23 (in Chinese with English abstract).
[16] Chen Z Y, Cheng X J, Chai L L, Wang Z H, Du D J, Wang Z H, Bian R L, Zhao A J, Xin M M, Guo W L, Hu Z R, Peng H R, Yao Y Y, Sun Q X, Ni Z F. Pleiotropic QTL influencing spikelet number and heading date in common wheat (L.)., 2020, 133: 1825–1838.
[17] 高德荣, 王慧, 刘巧, 朱冬梅, 张晓, 吕国锋, 张晓祥, 江伟, 李曼. 迟播早熟高产小麦新品种德选育. 中国农业科学, 2019, 52: 2379–2390. Gao D R, Wang H, Liu Q, Zhu D M, Zhang X, Lyu G F, Zhang X X, Jiang W, Li M. Breeding of new wheat varieties with early maturity and high yield under late sowing., 2019, 52: 2379–2390 (in Chinese with English abstract).
[18] 董玉琛. 小麦的基因源. 麦类作物学报, 2000, 20: 78–81. Dong Y C. Genepools of common on wheat., 2000, 20: 78–81 (in Chinese with English abstract).
[19] 张晓, 张伯桥, 江伟, 吕国锋, 张晓祥, 李曼, 高德荣. 扬麦系列品种品质性状相关基因的分子检测. 中国农业科学, 2015, 48: 3779–3793. Zhang X, Zhang B Q, Jiang W, Lyu G F, Zhang X X, Li M, Gao D R. Molecular detection for quality traits-related genes in Yangmai series wheat cultivars., 2015, 48: 3779–3793 (in Chinese with English abstract).
[20] 姜朋, 何漪, 张旭, 吴磊, 张平平, 马鸿翔. 宁麦9号与扬麦158株高及其构成因素的遗传解析. 作物学报, 2020, 46: 858–868. Jiang P, He Y, Zhang X, Wu L, Zhang P P, Ma H X. Genetic analysis of plant height and its commonents for wheat (L.) cultivars Ningmai 9 and Yangmai 158., 2020, 46: 858–868 (in Chinese with English abstract).
[21] 姜朋, 张旭, 吴磊, 何漪, 张平平, 马鸿翔, 孔令让. 宁麦9号/扬麦158重组自交系群体产量性状的遗传解析. 作物学报, 2021, 47: 869–881. Jiang P, Zhang X, Wu L, He Y, Zhang P P, Ma H X, Kong L R. Genetic analysis for yield related traits of wheat (L.) based on a recombinant inbred line population from Ningmai 9 and Yangmai 158., 2021, 47: 869–881 (in Chinese with English abstract).
[22] 何贤芳, 王晓波, 司红起, 夏云祥, 马传喜. STS标记在扬麦158×淮麦18 F4群体中的应用及其与PPO活性的关系. 分子植物育种, 2008, 6: 499–503. He X F, Wang X B, Si H Q, Xia Y X, Ma C X. The application of STS marker in Yangmai 158 × Huaimai 18 F4separating plant lines and its relations with PPO activity., 2008, 6: 499–503 (in Chinese with English abstract).
[23] 吴纪中, 蔡士宾, 颜伟, 任丽娟, 陈怀谷, 吴小有, 张仙义. ARz×扬麦158群体对小麦赤霉病抗性的QTL分析. 江苏农业学报, 2006, 22: 339–345. Wu J Z, Cai S B, Yan W, Ren L J, Chen H G, Wu X Y, Zhang X Y. QTL analysis of fusarium head blight resistance (FHB) in ARz×Yangmai 158 population., 2006, 22: 339–345 (in Chinese with English abstract).
[24] Wen M X, Su J X, Jiao C Z, Zhang X, Xu T, Wang T, Liu X X, Wang Z K, Sun L, Yuan C X, Wang H Y, Wang X E, Xiao J. Pleiotropic effect of the compactum gene and its combined effects with other loci for spike and grain-related traits in wheat., 2022, 11: 01837.
[25] Zhu T, Wang L, Rimbert H, Rodriguez J C, Deal K R, Deolivera R, Choulet F, Keeble-Gagnere G, Tibbits J, Rogers J, Eversole K, Appels R, Guo Y Q, Mascher M, Dvorak J, Luo M C. Optical maps refine the bread wheatcv. Chinese Spring genome assembly., 2021, 107: 303–314.
[26] Ma S W, Wang W, Wu J H, Guo W L, Chen Y M, Li G W, Wang Y P, Shi W M, Xia G M, Fu D L, Kang Z S, Ni F. WheatOmics: a platform combining multiple omics data to accelerate functional genomics studies in wheat., 2021, 14: 1965–1968.
[27] Fan X L, Cui F, Ji J, Zhang W, Zhao X Q, Liu J J, Meng D Y, Tong Y P, Wang T, Li J M. Dissection of pleiotropic QTL regions controlling wheat spike characteristics under different nitrogen treatments using traditional and conditional QTL mapping., 2019, 10: 187.
[28] Mizuno N, Nitta M, Sato K, Nasuda S. A wheat homologue ofis a candidate gene conferring the early heading phenotype to einkorn wheat., 2012, 87: 357–367.
[29] Zhang L, Zhang H, Qiao L Y, Miao L F,Yan D, Liu P, Zhao G Y, Jia J Z, Gao L F. Wheat MADS-box genenegatively regulates heading date., 2021, 9: 1115–1123.
[30] Zou J, Semagn K, Chen H, Iqbal M, Asif M, Ndiaye A, Navabi A, Perez-lara E, Pozniak C, Yang R C, Graf R J, Randhawa H R, Spaner D. Mapping of QTLs associated with resistance to common bunt, tan spot, leaf rust, and stripe rust in a spring wheat population., 2017, 37: 144.
[31] Griffiths S, Simmonds J, Leverington M, Wang Y K, Fish L, Sayers L, Alibert L, Orford S, Wingen L, Herry L, Faurer S, Laurie D, Biham L, Snape J. Meta-QTL analysis of the genetic control of ear emergence in elite European winter wheat germplasm., 2009, 119: 383–395.
[32] Hu J M, Wang X Q, Zhang G X, Jiang P, Chen W Y, Hao Y C, Ma X, Xu S S, Jia J Z, Kong L R, Wang H W. QTL mapping for yield-related traits in wheat based on four RIL population., 2020, 133: 917–933.
[33] Kane N A, Danyluk J, Tardif G, Quellet F, Laliberte J F, Limin A E, Fowler B, Sarhan F., a member of the StMADS-11 clade of flowering repressors, is regulated by vernalization and photoperiod in wheat., 2005, 138: 2354–2363.
[34] Mason R E, Hays D B, Mondal S, Ibrahim A M, Basnet B. QTL for yield, yield components and canopy temperature depression in wheat under late sown field conditions., 2013, 194: 243–259.
[35] Peng J, Richards D E, Hartley N M, Murphy G P, Devos K M, Flintham J E, Beales J, Fish L J, Worland A J, Pelica F, Sudhakar D, Christou P, Snape J W, Gale M D, Harberd N P. ‘Green revolution’ genes encode mutant gibberellin response modulators., 1999, 400: 256–261.
[36] Wang R X, Hai L, Zhang X Y, You G X, Yan C S, Xiao S H. QTL mapping for grain filling rate and yield-related traits in RILs of the Chinese winter wheat population Heshangmai × Yu 8679., 2009, 118: 313–325.
[37] Thambugala D, Brule-Babel A L, Blackwell B A, Fedak G, Foster A J, MacEachern D, Gillbert J, Henriquez M A, Martin R A, McCallum B D, Spaner D, Iqbal M, Pozniak C J, Diaye A N, McCartney C. Genetic analyses of native Fusarium head blight resistance in two spring wheat populations identifies QTL near the,,,,,, andloci., 2020, 133: 2775–2796.
[38] Addison C K, Mason R E, Brown-Guedira G, Guedira M, Hao Y F, Miller R G, Subramanian N, Lozada D N, Acuna A, Arguello M N, Johnson J W, Ibrahim A M H, Sutton R, Harrison S A. QTL and major genes influencing grain yield potential in soft red winter wheat adapted to the southern United States., 2016, 209: 665–677.
[39] Fatima S, Chaudhari S K, Akhtar S, Amjad M S, Akbar M, Iqbai M S, Arshad M, Shehzad T. Mapping QTLs for yield and yield components under drought stress in bread wheat (L.)., 2018, 16: 4431–4453.
[40] Zhuang M J, Li C N, Wang J Y, Mao X G, Li L, Yin J, Du Y, Wang X, Jing R L. The wheatgenecontrols root length in an auxin-dependent pathway., 2021, 72: 6977–6989.
[41] Singh K, Saini D K, Saripalli G, Batra R, Gautam T, Singh R, Pal S, Kumar M, Jan I, Singh S, Kumar A, Sharma H, Chaudhary J, Kumar K, Kumar S, Singh V K, Singh V P, Kumar D, Sharma S, Kumar S, Kumar R, Sharma S, Gaurav S S, Sharma P K, Balyan H S, Gupta P K. Wheat QTLdb V2.0: a supplement to the database for wheat QTL., 2022, 42: 56.
[42] 胡文静, 张勇, 陆成彬, 王凤菊, 刘金栋, 蒋正宁, 王金平, 朱展望, 徐小婷, 郝元峰, 何中虎, 高德荣. 小麦品种扬麦16赤霉病抗扩展QTL定位及分析. 作物学报, 2020, 46: 157–165. Hu W J, Zhang Y, Lu C B, Wang F J, Liu J D, Jiang Z N, Wang J P, Zhu Z W, Xu X T, Hao Y F, He Z H, Gao D R. Mapping and genetic analysis of QTLs for Fusarium head blight reistance to disease spread in Yangmai 16., 2020, 46: 157–165 (in Chinese with English abstract).
[43] Chen Z Y, Cheng X J, Chai L L, Wang Z H, Bian R L, Li J, Zhao A J, Xin M M, Guo W L, Hu Z R, Peng H R, Yao Y Y, Sun Q X, Ni Z F. Dissection of genetic factors underlying grain size and fine mapping ofD in common wheat (L.)., 2020, 133: 149–162.
[44] Lin Y, Jiang X J, Hu H Y, Zhou K Y, Wang Q, Yu S F, Yang X L, Wang Z Q, Wu F K, Liu S H, Li C X, Deng M, Ma J, Chen G D, Wei Y M, Zheng Y L, Liu Y X. QTL mapping for grain number per spikelet in wheat using a high-density genetic map., 2021, 9: 1108–1114.
Mapping and effect analysis of QTL for phenology traits in wheat using 55K chip technology
WEN Ming-Xing1,2,*, XIAO Jin2, XU Tao2, SUN Li2, WANG Zong-Kuan2, WANG Hai-Yan2, and WANG Xiu-E2
1Zhenjiang Institute of Agricultural Science, Jurong 212400, Jiangsu, China;2State Key Laboratory of Crop Genetics and Germplasm Enhancement / Nanjing Agricultural University, Nanjing 210095, Jiangsu, China
Phenology is animportant agronomic trait for common wheat, which has a great impact to explore its genetic mechanism and effect for wheat breeding and application. In this study, 240 recombinant inbred lines (RILs) derived from Yangmai 158 and Hiller were used to identify the phenology traits in 2–4 environments. A total of 52 QTL were detected on chromosomes 2A, 2D, 3D, 4B, 4D, 5A, 5B, 5D, 6A, 6B, 7A, 7B, and 7D by using the constructed high-density genetic map.,, andwere detected for several years, which explained 4.56%–46.86%, 1.32%–33.40%, and 2.37%–13.27% of phenotypic variation, respectively.-,-,,,,,,,,,,andwere new QTL.Pyramiding of multiple phenology loci with large effects or repeated is an effective approach to shorten the growth period in different degrees, which could be used to cultivate early-maturing and high-yield wheat varieties.
L.; phenology traits; QTL; SNP markers
2023-05-24;
2023-05-31.
10.3724/SP.J.1006.2023.31014
通信作者(Corresponding author): 温明星, E-mail: wmxcell2007@163.com
2023-02-26;
本研究由作物遗传与种质创新国家重点实验室开放基金项目(ZW20202009)资助。
This study was supported by the Open Fund of the State Key Laboratory of Crop Genetics and Germplasm Enhancement (ZW20202009).
URL: https://kns.cnki.net/kcms2/detail/11.1809.S.20230530.1546.008.html
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
