皱边喉毛花及其近缘种叶绿体基因组结构与进化分析
2023-11-14余静雅李孝平张发起
韩 霜,余静雅,李孝平,牛 玉,张发起
(1 中国科学院西北高原生物研究所高原生物适应与进化重点实验室,西宁 810001;2 中国科学院大学 生命科学学院,北京 100039)
基因组学与系统发育生物学相结合,建立新的学科——系统发育基因组学(Phylogenomics),也为正确解决多基因序列系统发育树冲突等问题提供新的方法和见解[1,2-4]。基因组包含丰富的基因序列信息及基因复制、转移、丢失等事件的重要信息,为系统发育研究提供大量证据。系统发育重建进入基因组时代,越来越多的学者将核基因组与质体基因组应用到系统发育学研究中[5-6],通过基因组数据构建更高分辨率、更成熟的系统发育树,进而更准确地推断植物类群的进化关系。对于高等植物和一些藻类植物而言,其体内固有的细胞器叶绿体在植物的光合作用过程中具有重要作用[7]。此外,由于叶绿体为母系遗传,高度保守、变异位点多并具有独立的基因组,是研究植物系统发育的理想目标[5]。测序技术的迅速发展与测序成本的降低,实现了许多植物的叶绿体基因组测序,为解决植物类群系统发育问题提供新的思路[8-9]。近年来,许多学者利用大量有价值的基因组数据解决了长期存在争议的植物系统发育问题。在这些研究案例中,对蔷薇科的123个叶绿体基因组进行测序,应用多种系统发育重建方法明晰蔷薇科的深层系统发育关系,为其多样化历史提供了新的重要见解[10]。对麻黄属3个物种的叶绿体基因组进行测序,为其系统发育研究提供有价值的信息[11]。
喉毛花属(Comastoma)隶属于龙胆科(Gentianaceae)獐牙菜亚族(subtribe Swertiinae),多为一年生或多年生草本植物,全球共15种,中国有11种[12]。该属植物主要分布在青藏高原及其毗邻地区,也是藏茵陈入药基原植物之一,在保肝、抗氧化、抗病毒等方面具有显著效果[13-15]。综合以往研究分析,学者基于ITS序列对龙胆亚族进行系统发育分析,支持喉毛花属为单系起源,胚胎学研究将喉毛花属处理为1个独立的属,质体基因组与核基因组研究结果进一步支持该属为1个单系类群[16-20]。目前,喉毛花属属级分类问题已明晰,但其属下物种关系问题尚未得到解决。此外,研究者还发现不同数据集构建的系统发育树表现出不一致的拓扑结构[16,20],主要表现在皱边喉毛花(Comastomapolycladum)、镰萼喉毛花(Comastomafalcatum)等物种上。目前,喉毛花属下物种关系尚不清晰,有关喉毛花属下物种的系统发育研究鲜见报道,对其属下物种的认识有限。因此,后续研究应结合更多丰富、有价值的基因组数据阐明喉毛花属下物种分类问题。鉴此,文章以皱边喉毛花及其近缘种作为研究对象,通过生物信息学方法进行基因组比较、进化与密码子偏好性等分析,构建不同数据集,重建喉毛花属下物种的系统发育关系,为进一步展开喉毛花属下物种分化研究提供可靠的分子依据。
1 材料和方法
1.1 材 料
选取12个喉毛花属植物样本(5个皱边喉毛花、2个长梗喉毛花、2个镰萼喉毛花和2个高杯喉毛花),这些样本分别采自不同地区与不同群体(表1)。野外采集的新鲜幼叶放入硅胶中干燥,回到实验室后,置于-20 ℃冰箱保存。凭证标本存放于中国科学院西北高原生物研究所青藏高原生物标本馆(HNWP)内。
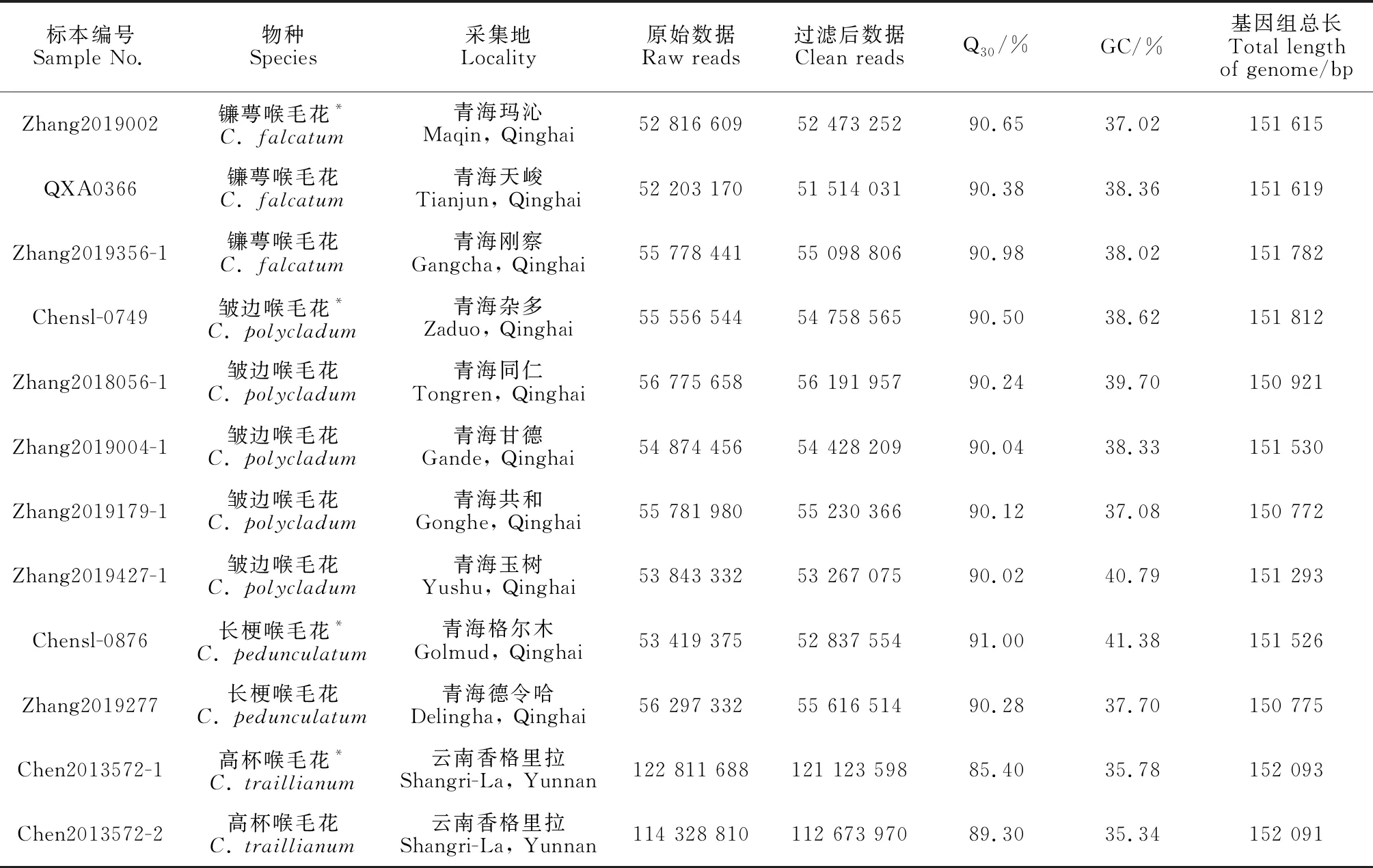
表1 物种信息与测序结果
1.2 DNA提取与测序
采用改良后的CTAB法从干燥叶片中提取基因组总DNA[21],琼脂糖凝胶电泳检测DNA纯度和完整性,送至公司构建全基因组小片段文库。构建完成后使用Agilent 2100对文库插入片段进行检测。文库检测合格后,利用Illumina高通量测序平台NovaSeq 6000平台进行150 bp长度的双末端测序。
1.3 叶绿体基因组组装与注释
利用fastp v 0.23.1[22]对原始数据(raw reads)进行质量控制和过滤(参数设置:-poly_x_min_len:10;cut_front cut_tail cut_window_size:4;qualified_quality_phred:15;low_complexity_filter complexity_threshold:30;length_required:30 thread:4),过滤后的高质量数据(clean reads)用于后续生物信息学分析。使用SPAdes v 3.13.1 与Getorganelle v 1.7.7.0组装12个个体的叶绿体基因组[23-24],提交至在线工具Geseq进行基因组注释,得到Genbank文件[25],随后提交至Sequin软件,手动去除内含子及外显子,调整起始/终止密码子的位置,最终导出用于后续分析的Genbank文件并提交至NCBI数据库中。此外,从本数据库中下载已经发表的龙胆科4个属獐牙菜属(Swertia)、花锚属(Halenia)、扁蕾属(Gentianopsis)与肋柱花属(Lomatogonium)的9个物种:蒙自獐牙菜(Swertialeducii,登录号:NC_045301,153 015 bp)、显脉獐牙菜(Swertianervosa,登录号:NC_057596,153 690 bp)、祁连獐牙菜(Swertiaprzewalskii,登录号:MZ261904,153 269 bp)、宿根肋柱花(Lomatogoniumperenne,登录号:NC_050659,151 678 bp)、椭圆叶花锚(Haleniaelliptica,登录号:NC_050657,153 305 bp)、Haleniacoreana(登录号:NC_050657,NC_042674,153 198 bp)、大花扁蕾(Gentianopsisgrandis,登录号:NC_049879,151 271 bp)、湿生扁蕾(Gentianopsispaludosa,登录号:NC_050656,151 308 bp)和扁蕾(Gentianopsisbarbata,登录号:MZ579704,151 123 bp),獐牙菜属物种作为外类群。
1.4 密码子偏好性分析
选取4个同科植物(显脉獐牙菜、宿根肋柱花、扁蕾与椭圆叶花锚)与1个已发表的喉毛花MK331815,与皱边喉毛花(Chensl-0749)、镰萼喉毛花(Zhang2019002)、长梗喉毛花(Chensl-0876)和高杯喉毛花(Chen2013572-1)进行密码子偏好性分析。使用codonW v 1.4.2(http://codonw.sourceforge.net/)计算相对同义密码子使用度(relative synonymous codon usage,RSCU)、密码子适应指数(codon adaptation index,CAI)、有效密码子数(effective number of codons,ENc)、同义密码子第三位GC含量(GC content of the synonymous third codons,GC3s)、同义第三密码子胸腺嘧啶含量(synonymous third codon thymine content,T3s)、同义第三密码子胞嘧啶含量(synonymous third codon cytosine content,C3s)、同义第三密码子腺嘌呤含量(synonymous third codon adenosine content,A3s)和同义第三密码子鸟嘌呤含量(homonymous third codon guanine content,G3s)等指数。在TBtools v 1.09中绘制密码子偏好性分析图[26]。
1.5 叶绿体基因组比较分析
为比较喉毛花属及其近缘属的叶绿体基因组相似性及差异性,利用mVISTA软件[27]进行可视化分析。以镰萼喉毛花作为参考,与其余8个物种(显脉獐牙菜、宿根肋柱花、扁蕾、椭圆叶花锚、皱边喉毛花、长梗喉毛花、喉毛花及高杯喉毛花)进行比较,选择Shuffle-LAGAN模型,其他参数设为默认值。利用MISA v 1.0[28]进行短重复序列(short repeat sequence)分析,参数设置:1=10;2=5;3=4;4=3;5=3;6=3,利用Excel 2016绘制SSR数量及其分布情况。
在Phylosuite v 1.2.2[29]中提取9个GB文件的编码基因(protein-coding gene,PCGs)以及基因间隔区(intergentic spacers,IGS),在MAFFT v 7.313[30]中进行基因及基因间隔区的序列比对,得到的比对结果为输入文件进行核苷酸多态性分析。将提取的共有基因分为不同的数据集:accD(fatty acid synthesis)、atp(ATP synthase)、cemA(carbon metabolism)、clpP(proteolysis)、infA(translational initiation factor)、matK(RNA processing)、ndh(NADPH dehydrogenase)、pet(cytochrome b/f complex)、psa(photosystem I)、psb(photosystem II)、rbcL(rubisco)、rpl(large subunit of ribosome)、rpo(DNA dependent RNA polymera)、rps(small subunit of ribosome)。利用DNAsp v 6[31]的Nucleotide diversity模块计算PCGs和IGS的Pi值(参数设置:滑动窗口大小为600 bp,为200 bp),利用Excel 2016绘制结果图。从结果中筛选高度变异基因间隔区(Pi>0.06)用于系统发育分析。
以显脉獐牙菜为参考,利用软件KaKs_Calculator v 2.0[32]计算14个数据集的同义替代率(synonymous,Ks)、非同义替代率(nonsynonymous,Ka)及同义替代率/非同义替代率的比值(Ka/Ks)(method:NG;genetic code:11-Bacterial and plant plastid code)。在R v 4.2.4(https://www.R-project.org/)中利用ggplot2包绘制进化分析图。
1.6 喉毛花属下的系统发育分析
共构建了4个数据集:蛋白编码序列(CDS),第一、二位密码子位置(CDS1+2),第三位密码子位置(CDS3)以及筛选出的高度变异基因间隔区(ndhC_trnV-UAC、psbC_trnS-UGA和psbK_psbI)序列用于喉毛花属下系统发育关系重建。利用MAFFT v 7.313[30]对共有CDS基因以及高度变异IGS进行比对(密码子比对策略),对24条叶绿体基因组序列共有的82个编码基因进行多基因联合分析。CDS基因的密码子位点数据集采用Partition估算建树最佳模型,其余数据集在Modelfinder中计算最佳模型[33-34](表2)。在IQ-tree v 1.6.8[35]中进行最大似然(maximum likelihood,ML)分析,选择Ultrafast bootstrap,快速自然重复设置为1 000次。使用MrBayes v 3.2.6[36]进行贝叶斯(bayesian inference,BI)分析,每次分析进行2次独立的马尔可夫链蒙特卡罗(MCMC)链运行,迭代选择1 000 000代,每1 000代选1次样,舍弃前25%的样本。利用ITOL(interactive tree of life, https://itol.embl.de/)在线软件对结果图进行可视化和修改。

表2 所有数据集的最佳建树模型
2 结果与分析
2.1 叶绿体基因组结构特征
经测序后,对12个样本的测序质量进行评估,其中,10个样本的Q30均在90%以上(除高杯喉毛花的2个样本外),GC含量范围在35.34%~41.38%之间,基因组长度范围在150 772~152 093 bp之间(表1)。经组装及注释后,获得12个完整的叶绿体基因组,均由四分体结构组成,包括大单拷贝区(large single copy,LSC)、小单拷贝区(small single copy,SSC)及1对反向重复区(inverted repeat region,IRa、IRb)。选取9个叶绿体基因组进行比较分析,叶绿体基因组长度范围在151 123~153 690 bp之间(表3)。
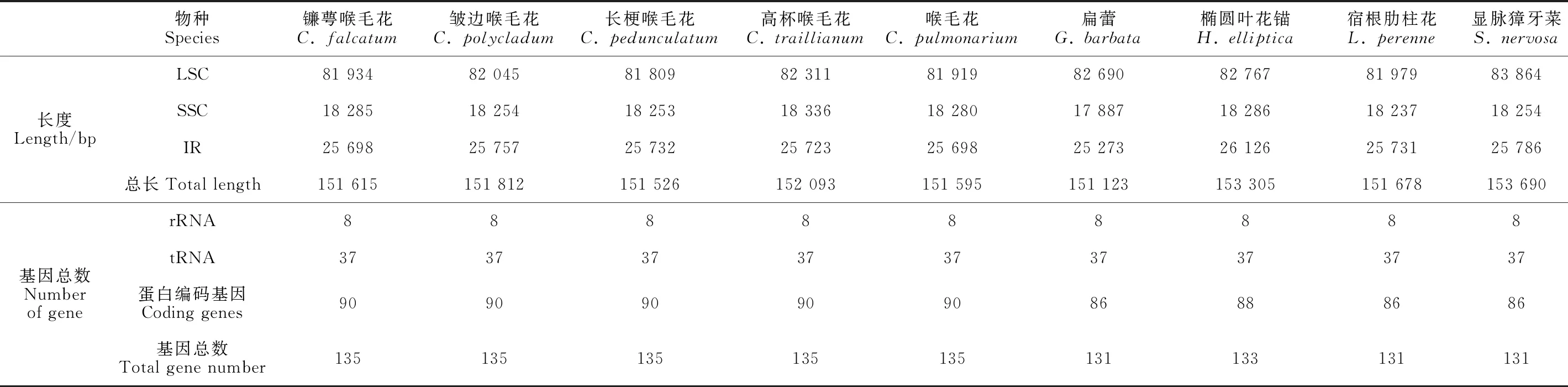
表3 9个叶绿体基因组结构信息
2.2 密码子偏好性分析
基于82个共有基因对喉毛花属物种的密码子偏好性进行分析,结果显示,喉毛花属与其近缘属之间略有差异,主要体现在编码蛋白基因的密码子数量不同,范围为50 374~51 230。在20个氨基酸中,亮氨酸(Leu)具有最多的密码子,而色氨酸(Trp)具有最少的密码子。RSCU结果表明,有35个密码子的RSCU值均大于1,其余密码子均≤1(图1)。密码子指数计算结果显示,所有物种的GC含量相差不大,均在0.39~0.394之间。密码子适应指数CAI在0.15~0.162之间,有效密码子数目ENc值在55.89~56.44之间(表4)。
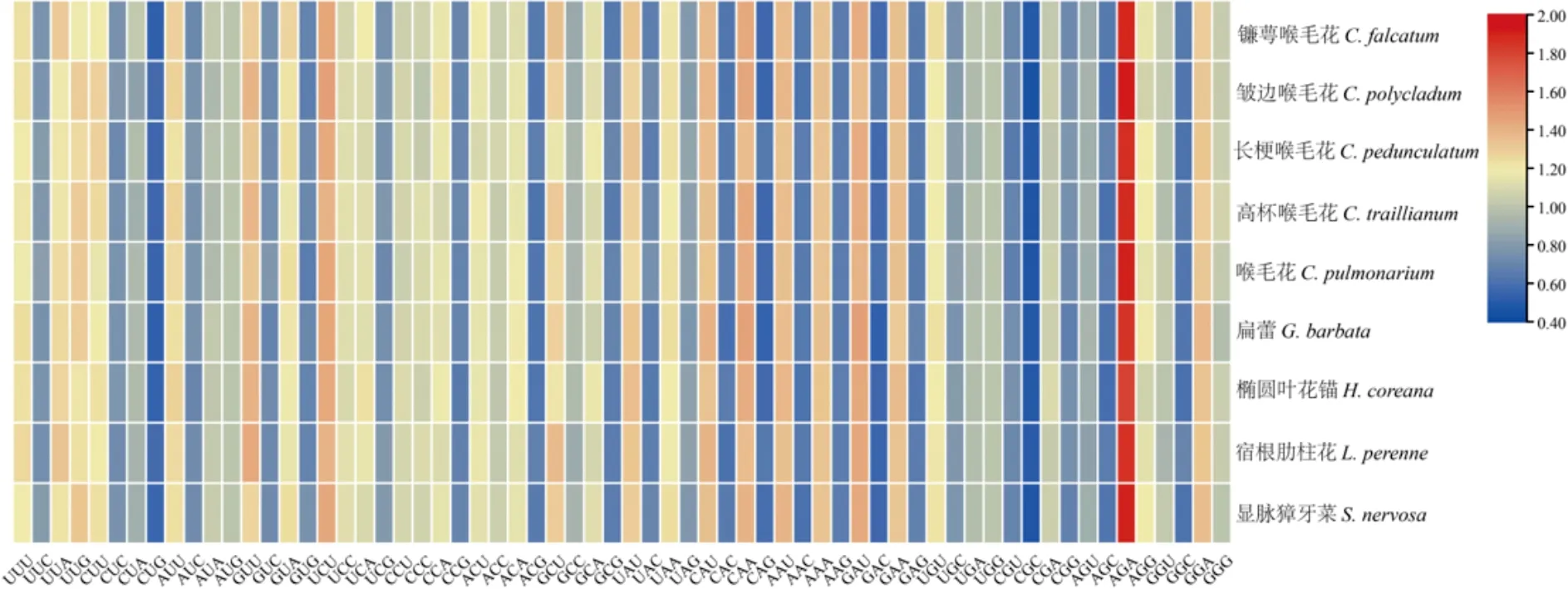
图1 9个叶绿体基因组的密码子偏好性热图
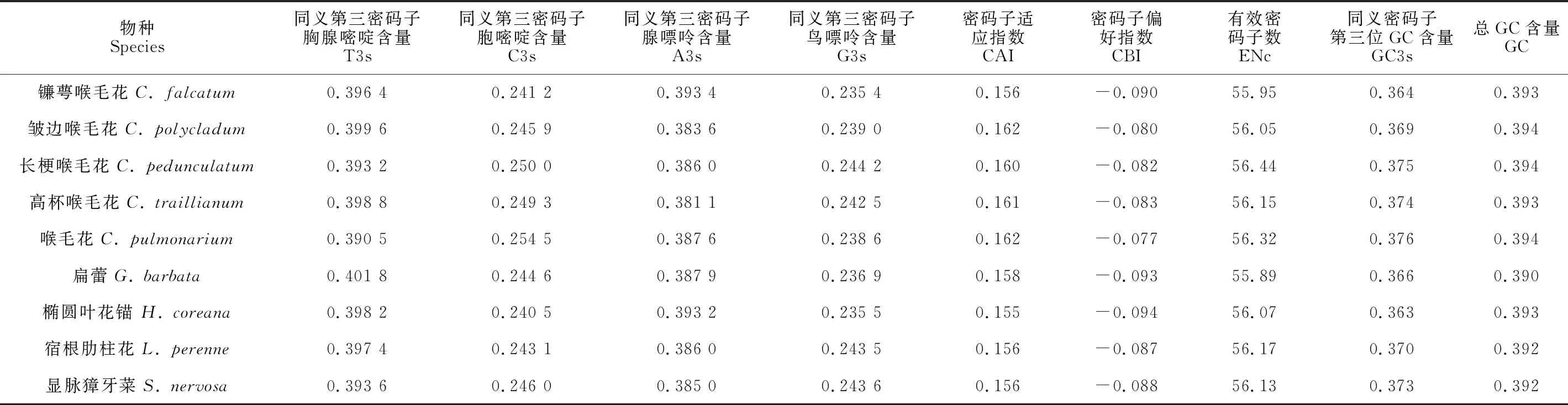
表4 叶绿体基因组编码基因的密码子偏好性指数
2.3 叶绿体基因组比较分析
9个叶绿体基因组序列相似性可视化分析显示编码区比非编码区更保守,相比于反向重复区单拷贝区差异性更大,与核苷酸多态性分析结果基本保持一致(图2)。SSR分析结果显示,喉毛花属及其近缘属中的重复类型总数在42~53之间,其中扁蕾和显脉獐牙菜的数量最多。单碱基重复类型最丰富,范围在28~37之间(图3)。在10个基因数据集的核苷酸多态性结果中,其Pi值范围为0.005~0.037。其中,cemA、matK基因具有较高的Pi值,说明它们在喉毛花属植物中的变异率最高,而基因间隔区的结果中,笔者只展示Pi值大于0.05的基因间隔区,并选取Pi大于0.06的基因间隔区作为数据集构建系统发育树(图4)。

图2 喉毛花属及其近缘类群的叶绿体基因组比较
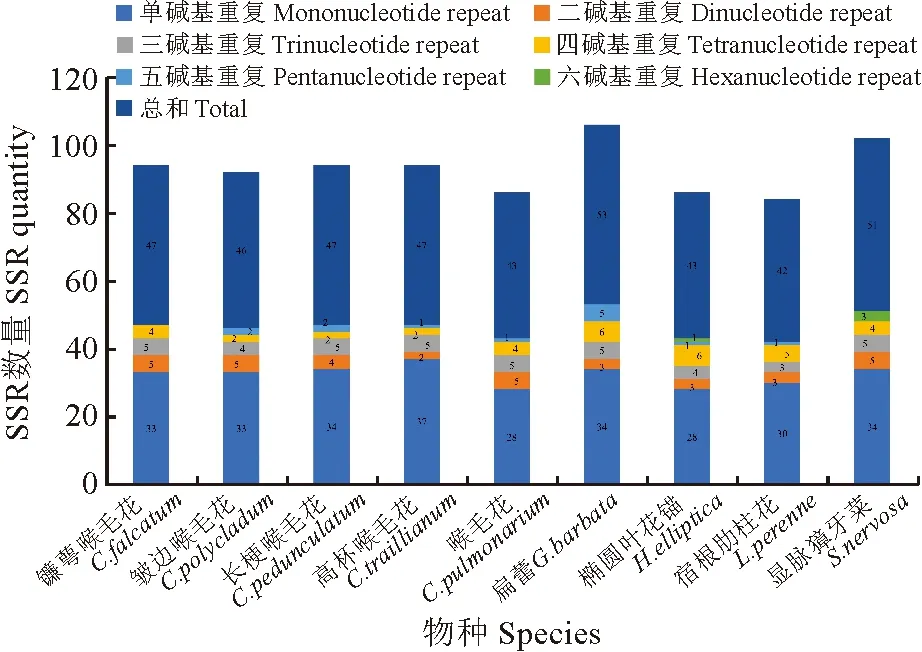
图3 9个叶绿体基因组分SSR分析
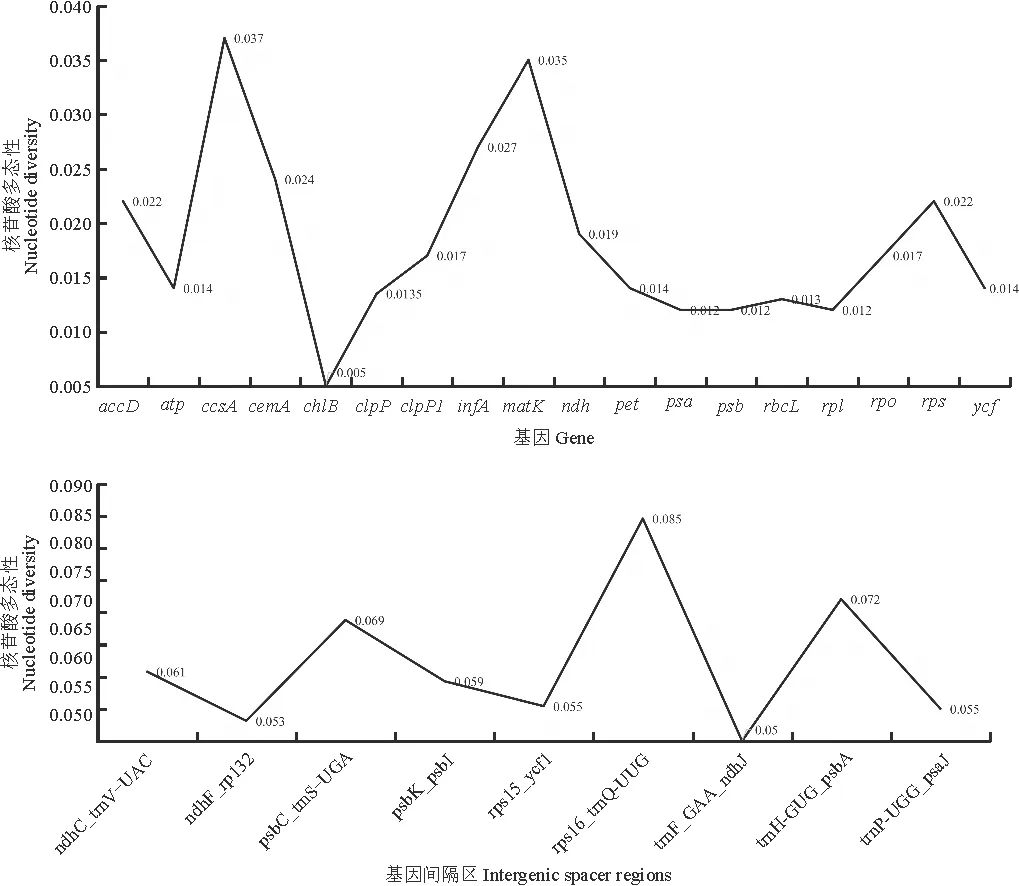
图4 蛋白编码基因和基因间隔区的核苷酸多态性分析
2.4 叶绿体基因组进化分析
为进一步检测叶绿体基因组编码基因受到的选择压力,利用9个叶绿体基因组的共有编码基因,将其分为不同的9个基因数据集,计算同义替代率、非同义替代率以及Ka/Ks值(图5)。结果表明,非同义替换率均较低,范围在0.002~0.035之间,而同义替代率的范围值在0.003~0.11之间。非同义替代率与同义替代率的比值(Ka/Ks)结果中,psbA基因拥有最小值0.005,而ndhB基因的比值(1.24)最大。

图5 蛋白编码基因的同义替代率、非同义替代率以及Ka/Ks比率
2.5 喉毛花属下的系统发育分析
基于CDS、密码子第一、二位置及第三位置构建的ML和BI系统发育树具有相似的拓扑结构,所有分支具有较高的支持率(图6)。从系统发育树可知,所有的喉毛花属物种均聚为一支,其姊妹类群为肋柱花属植物,二者共同与花锚属的3个物种和獐牙菜属的1个物种聚为1个大支。而基因间隔区的系统发育树结果也具有相似的结果(图7)。

图6 基于叶绿体基因组蛋白编码序列,第1、2位密码子位置与第三位密码子位置的喉毛花属下系统发育关系

图7 基于高度变异基因间隔区联合喉毛花属下系统发育关系
从喉毛花属的分支结果可知,皱边喉毛花、镰萼喉毛花、长梗喉毛花均聚在了同一支上且相互嵌套在一起。在分支A中,2个镰萼喉毛花(QXA0366和Zhang2019356-1)与1个皱边喉毛花(Chensl-0749)聚在一支(分支a1),与分支b1构成姐妹类群。在分支b1中,皱边喉毛花(Zhang2019427-1)与镰萼喉毛花(MK331815)、长梗喉毛花(Zhang2019277)和皱边喉毛花(Zhang2019004-1)构成姐妹类群。分支B由分支a2和b2构成,二者互为姐妹类群,在分支a2中,皱边喉毛花(Zhang2018056)与皱边喉毛花(Zhang2019179-1)、长梗喉毛花(Chensl-0876)聚为一支。在分支b2中,2个高杯喉毛花(Chen2013572-1/2)与镰萼喉毛花(Zhang2019002)、2个喉毛花(NC_050654、MW324577)互为姐妹类群。基于以上结果可以得知,皱边喉毛花及其近缘种相互嵌套,未按物种聚类。
此外,联合变异程度较高的基因间隔区(ndhC_trnV-UAC、psbC_trnS-UGA和psbK_psbI)构建了ML和BI树,拓扑结构进一步说明喉毛花属的单系性(图7)。在2种系统发育树中,部分物种表现出不一致的拓扑结构。
在最大似然树中,长梗喉毛花(Chensl-0876)与皱边喉毛花(Zhang2018056)互为姐妹类群,而在贝叶斯树中,其与皱边喉毛花(Zhang2019004-1)聚在一起(图7)。类似的结果进一步说明皱边喉毛花、镰萼喉毛花与长梗喉毛花无法按照物种聚类,与CDS系统发育树结果一致。
3 讨 论
测序技术的迅速发展与测序成本的降低,实现了许多植物的叶绿体基因组测序,其长期作为解决植物类群系统发育问题的有利手段[8-9,37]。对于系统发育位置存在争议的喉毛花属而言,叶绿体基因组数据构建出成熟、高分辨率的喉毛花属系统发育树,支持了该属为单系起源。综合历史研究分析,喉毛花属的系统发育位置问题已得到解决,但其属下物种的系统发育关系尚不明晰[18]。一些系统发育研究案例中,不同数据构建的系统发育树表现出不一致的拓扑结构,类似情况也在其他植物类群中发现[38-40]。为此,本研究构建了不同数据集重建喉毛花属下的系统发育关系,以期为喉毛花属下物种关系的研究提供有价值的分子信息。
对测序原始数据的质控结果显示,其Q20均在96%以上,Q30值基本在90%以上,进一步说明其测序质量良好,可以进行后续的生物信息学分析。组装及注释后的叶绿体基因组为四分体结构,均由1个LSC区、SSC区和1对IR区组成。5个喉毛花属物种在基因组长度差异较小,长度范围为从151 526~152 093 bp不等,长梗喉毛花物种的基因组长度最短。同属物种之间的基因组长度差异是叶绿体基因组研究中最常见的结果,类似的结果也在其他植物中发现[41-42]。这种差异主要与某些基因的扩张和收缩有关,从而影响基因组大小[43]。
叶绿体基因组比较分析表明,喉毛花属下物种之间的差异较小。mVISTA以及核苷酸多态性结果表明,非编码区比编码区的变异程度高,反向重复区比单拷贝区的核苷酸多态性低,这与大多数植物的结果[44-45]保持一致。此外,筛选出变异程度高的基因间隔区或基因可作为进一步研究喉毛花属物种鉴定及群体遗传的分子标记候选。SSR结果表明,单碱基重复最为丰富。这与其他青藏高原地区的植物结果保持一致,也与多数植物的结果不一致,或与SSR分析所设定的参数、测序方法及物种本身有关[46]。这些结果可为进一步设计SSR引物及开发高度多态性的SSR标记提供基础数据。
同义核苷酸替换率和非同义核苷酸替换率的计算,对于植物系统发育的重建具有重要意义,可以判断植物叶绿体基因组编码基因是否受到选择压力的作用[47-48]。同义核苷酸的替代并不改变蛋白质的结构,而非同义核苷酸的替代却能改变蛋白质结构,因此非同义替换往往带来有害性状而被固定下来[49]。当Ks=Ka,说明编码基因未受到自然选择的影响[50];如果Ka/Ks>1,说明基因受正选择的作用。喉毛花属植物的进化分析结果显示,几乎所有编码基因的非同义替代率与同义替代率比值(Ka/Ks)均小于1,表明Ks>Ka,进一步说明这些编码基因受到纯化选择的作用。以上结果与其他植物的结果[11,50]保持一致。本研究筛选的正选择基因,能够为进一步研究喉毛花属下物种的适应性进化提供基础数据。
密码子偏好性分析结果表明喉毛花属的20个氨基酸均由61个密码子编码(3个终止密码子除外)。计算多个衡量指标可以量化植物叶绿体基因组密码子的偏好性,RSCU可以很直观地展示密码子使用的偏好性[51]。在喉毛花属植物中,有1/2以上密码子的RSCU值大于1,说明使用这些密码子的频率较高。ENc值可以反映出同义密码子非均衡使用的偏好程度,其结果具有较高的参考价值[52]。当ENc值越小时,其密码子的偏好程度高。因此,通过比较ENc值可以确定内源基因表达量的相对高低[52]。本研究中,喉毛花属物种的ENc范围在55~57之间(均大于35),说明该属物种的叶绿体基因密码子偏好性较弱。
喉毛花属下物种系统发育关系重建结果显示,基于CDS、密码子位置与基因间隔区数据集的系统发育树均表现出高度一致的拓扑结构。该结果进一步支持喉毛花属植物为单系类群,肋柱花属植物为该属的近缘类群,与之前的研究结果[18]保持一致。在支持率良好的喉毛花属下的系统发育关系结果中,皱边喉毛花、长梗喉毛花和镰萼喉毛花各自并未按物种进行聚类,而是相互嵌套,在联合IGS区构建的系统发育树中也出现类似结果,进一步支持ITS、matK基因系统发育树的结果[20]。利用质体基因组数据重建喉毛花属下物种系统发育关系的研究受到限制,无法明晰皱边喉毛花及其近缘种之间的关系。在后续的研究中,需要对这些物种展开群体采样并进行物种分化研究。
本研究利用二代测序技术对皱边喉毛花及其近缘物种进行了叶绿体基因组的测序与组装,获得的测序数据用于分析叶绿体基因组比较、进化、密码子偏好性等分析,并基于不同数据集和变异程度较高的基因间隔区联合进行系统发育树的构建,所呈现的系统发育拓扑结构一致。然而,皱边喉毛花及其近缘物种未按物种分类,各自并未形成独立的分支。对于喉毛花属下的物种分化问题需要开展进一步的群体遗传学研究,基于现有的成熟技术,揭示属下物种关系,阐明物种形成机制。
