桤木Genomic-SSR 与EST-SSR 分子标记的差异分析
2023-11-13王泽亮杨勇智杜晋城陈炙黄振郭洪英
王泽亮 ,杨勇智,杜晋城,陈炙,黄振,郭洪英,2
1.四川省林业科学研究院,四川 成都,610081;2.四川省草原科学研究院,四川 成都, 611731
桤木属(AlnusMill.)为非豆科固氮树种,根系富含根瘤,可改良土壤,是重要的先锋造林与生态功能树种。桤木属是现存桦木科植物中最原始的属,也是北半球新生代植物区系的重要植物类群,主要分布在欧亚和北美,拉丁美洲与非洲有少量分布[1-2]。四川省及邻近地区是桤木属的重要分布区,原生分布有桤木(Alnus cremastogyne,又名四川桤木)、川滇桤木(A.ferdinandi-coburgii)、毛桤木(A.lanata)、尼泊尔桤木(A.nepalensis),可能是桤木属植物起源与分化的中心[1],其中桤木是我国最重要的一个特有种,也是研究最广泛的一个种,生长迅速、适应性强、童期短且结实量大,目前其适生栽培区域已扩大至长江中下游地区,是中国长江流域退耕还林工程、生态建设工程和混交造林的重要树种。
SSR(Simple sequence repeat,简单序列重复)分子标记具有分布广泛、多态性丰富、稳定、共显性等特点,自从被开发以来,在物种遗传改良上获得了广泛的应用。在桤木属植物SSR 研究方面,Zhuk 等最早开发出了桤木SSR 引物[3]。随后,Lance等通过筛选海岸桤木(A.maritima)基因组文库获得了19 条桤木SSR 引物[4]。使用Lance 开发的引物,Jones 等人系统研究了美国濒危物种海岸桤木的遗传多样性与群体结构等,为其提供了的濒危保护理论基础[5,6]。随后SSR 技术逐渐的应用到其他桤木属树种中,这些树种的群体遗传变异基础与进化史也逐渐被深入揭示[7-10]。目前国内桤木遗传改良研究主要集中于育苗、栽培等常规育种方面,在群体遗传变异研究上,主要通过表型鉴定方法进行[11-14],采用分子标记手段的研究较少,仅有卓仁英等建立了RAPD 体系[15]。在SSR 分子标记方面,也仅有饶龙兵等基于桤木、欧洲桤木(A.glutinosa)、硬桤木(A.firma)转录组数据开发了适用于桤木属的SSR标记[16]。总之桤木群体遗传变异缺乏分子水平上的数据支撑,影响了其保护与进一步的推广利用。
此外,SSR 分子标记从来源上说包括Genomic-SSR 与EST-SSR,分别来源于基因组数据与表达序列标签(Express sequence tags,EST)数据。本研究分析了2 种来源的桤木SSR 标记的差异,以期推动其在国内桤木遗传变异研究上的应用。
1 材料与方法
1.1 植物材料
采样群体为天然次生林,共包括13 个群体(表1)。群体范围包括成都平原区、盆周山地区、盆地丘陵区、川西高山峡谷区和川西南山地区。群体取样单株之间相距至少50m,采集桤木新鲜叶片,硅胶干燥保存带回实验室。
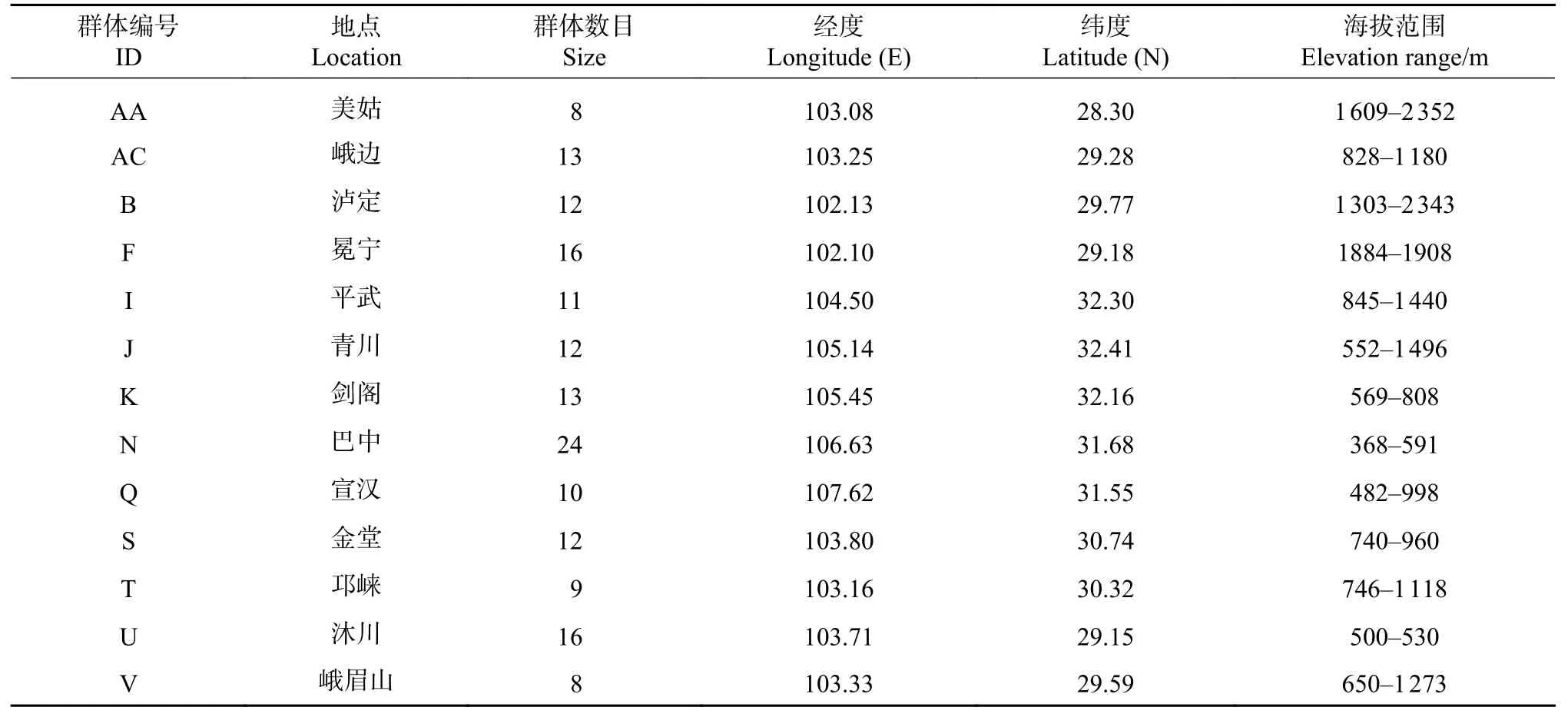
表1 桤木群体基本信息Tab.1 Location and number of trees sampled in 13 Alnus Cremastogyne populations
1.2 DNA 提取与PCR 扩增
使用天根植物基因组提取试剂盒DNA(DP305)提取基因组DNA。
SSR 扩增所用引物来源于2 部分:(1)Genomic-SSR,公开发表文献中其它桤木属树种相关研究中使用的SSR 引物[3-4,17-18],10 对引物;(2)EST-SSR[19],6 对引物。具体信息见表2。
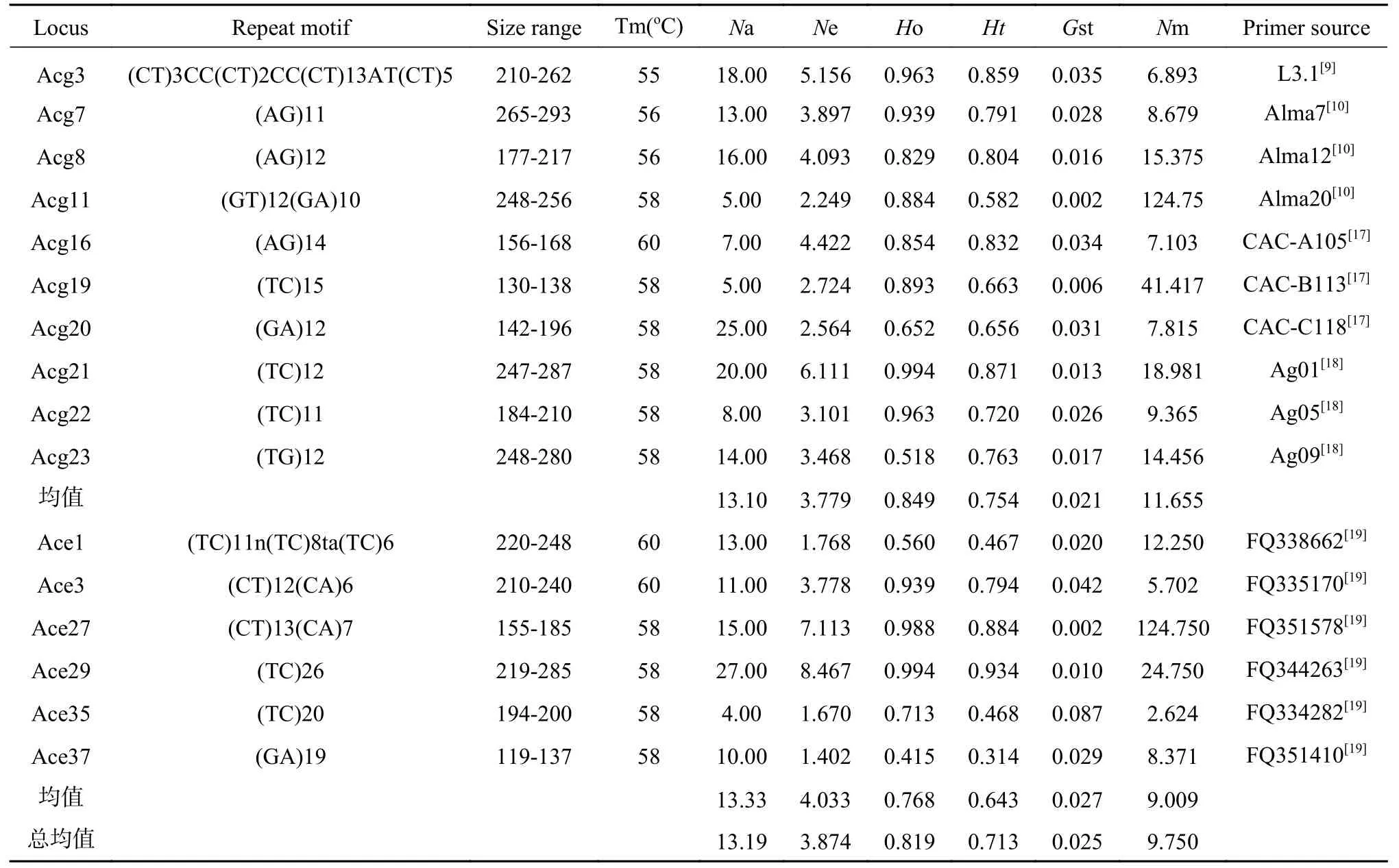
表2 桤木10 个Genomic-SSR 与6 个EST-SSR 标记遗传参数Tab.2 Genetic diversity of 164 trees in A.cremastogyne revealed by 10 Genomic-SSRs and 6 EST-SSRs
SSR 上游引物添加FAM 荧光标记。PCR 扩增使用Takara Taq 聚合酶(Takara,Dalian,China),20 uL反应体系包括:13.85 μL ddH2O,2.0 μL 10 × buffer,2.0 μL 2.0 mM dNTP,0.5 μL of each primer (at 10 μM),1 μL 基因组DNA 模版(30-50ng),and 0.75 U Taq DNA 聚合酶。扩增程序如下:94 ℃预变性5 min;94 ℃变性20 s,退火温度退火20 s,72 ℃延伸40 s,25-30 个循环;72 ℃延长5 min,4 ℃保存。退火温度根据文献或引物设计软件给出的数据。PCR 产物进行毛细管荧光电泳分型,SSR 片段长度由软件GeneMarker version 2.2.0 (SoftGenetics,USA)判读。
1.3 SSR 数据统计分析
首先利用Excel2007 整理整合GeneMarker 输出的基因型分子量数据,随后数据输入基于R 环境的Polysat 1.6 软件[20],整合相关信息后,最后输出为GenoDive 格式文件进行进一步分析。利用GenoDive 2.0b27[21]计算等位基因数(Na)、有效等位基因数(Ne)、观测杂合度(Ho)、总望杂合度(Ht)、固定系数(Gst,同Fst)等遗传参数,评价各群体的遗传多样性水平。基因流值(Nm)根据公式(1-GST)/(4GST)估算[22]。
利用GenoDive 2.0b27 计算Nei’s(1978)遗传距离,运用NTSYS-pc 2.10s 软件生成UPGMA 聚类图并计算各遗传距离的相关系数[23-24]。
2 结果与分析
2.1 桤木Genomic-SSR 与EST-SSR 多态性比较
利用10 个Genomic-SSR 位点与6 个EST-SSR个位点检测了13 个桤木群体164 个个体的遗传多样性参数,16 个位点全部具有多态性。进一步分析结果表明(表2),在10 个Genomic-SSR 位点中,平均等位基因数为13.10,平均有效等位基因数为3.779,平均观察杂合度为0.849,平均期望杂合度为0.754;对EST-SSR 来说,平均等位基因数为13.33,平均有效等位基因数为4.033,平均观察杂合度为0.768,平均期望杂合度为0.643。平均等位基因数EST-SSR 位点高于Genomic-SSR 位点,而对检测的杂合度来说,Genomic-SSR 位点高于EST-SSR 位点。就单个位点而言,揭示遗传多性度最高的为EST-SSR 位点Ace29,其等位基因数达到27,有效等位基因数为8.467,观察杂合度为0.994。在10 个基因组SSR 位点中,等位基因数≥20 的位点有2 个,在10 与20 之间的位点有4 个,10 以下的位点有4 个;而6 个EST-SSR 位点中,有1 个超过20,在10(包括等于)与20 之间的位点有4 个,10 以下的位点有1 个。
针对13 个桤木群体来说,除了平均有效等位基因数 EST-SSR 位点高于 Genomic-SSR(4.251>3.941)之外,其余3 个遗传参数(平均等位基因数、观察杂合度、期望杂合度)Genomic-SSR 位点均高于EST-SSR。对于具体群体来说,Genomic-SSR 与EST-SSR 位点分析均显示F(冕宁)群体遗传多样性水平最高,但是Genomic-SSR 位点显示I(平武、Ne 值最小)、J(青川、Na 与Ho 值最小)、Q(宣汉、Ht 值最小)群体遗传多样性水平最低,EST-SSR 位点显示I(平武、Na、Ne、Ho 值最小)、U(沐川、Ht 值最小)群体遗传多样性水平最低。
2.2 聚类分析
为确定两种标记对桤木群体遗传关系的鉴定准确度,本研究基于Nei’s 遗传距离使用UPGMA 方法对参试材料进行聚类分析,由图1(A、B)可知,Genomic-SSR 与EST-SSR 均将AA 与F 群体归为1 类,进一步Genomic-SSR 标记将其余群体归为3 类:AC、B;I、S、T、V;J、Q、U、K、N,而EST-SSR 划分的3 类为:AC;B、T;I、J、K、N、Q、U、V、S。说明在大的区域分类上,Genomic-SSR 与EST-SSR 相一致,而在小的分类上有差异。

图1 基于Nei’s 遗传距离的桤木群体UPGMA 聚类Fig.1 Unweighted pair-group method with arithmetic means (UPGMA) cluster analysis of 13 A.cremastogyne populations based on Nei’s genetic distance (A:Genomic-SSR;B:EST-SSR;C:Genomic-SSR+EST-SSR)
基于Genomic-SSR 与EST-SSR 总的16 个标记的Nei’s 遗传距离构建的UPGMA 聚类树显示桤木群体可明显地分为4 个类群(AA 与F、B、I、其它)(图1 C),与Genomic-SSR 标记结果更为相似,说明本研究中Genomic-SSR 更能精确地鉴别出桤木群体遗传关系。
对 Genomic-SSR、EST-SSR 以及 Genomic-SSR+EST-SSR 计算出的遗传距离进行相关性分析,结果显示(表3):3 部分相关系数呈极显著正相关(P<0.01),但是Genomic-SSR 与Genomic-SSR+EST-SSR 相关系数稍高,即两种标记计算的综合相关系数与Genomic-SSR 标记更为相似,表明本研究中Genomic-SSR 能更准确地揭示不同桤木个体间的遗传关系,与上述聚类分析结果相一致。
3 讨论
3.1 桤木倍性
在相关研究中,欧洲桤木是被作为二倍体树种进行研究的,随后Lepais 等[7]与Mandák 等[25]在非洲与欧洲分别发现了四倍体群体(2n=4x=56)。在染色体水平上,任保青等[26]与杨汉波等[27]通过核型研究发现桤木染色体数为56,根据洪德元提到桤木属的染色体基数为x=14[28],或Murai 认为的X=7[29],则桤木可能是四倍体或八倍体。而核型分析显示桤木染色体结构相同的染色体对数多数为2 对[27],表明桤木可能正在进行或接近完成二倍体化。由于本研究种桤木SSR 分型数据表现出了四倍体特性,因此本研究以多倍体分析软件Polysat、Genodive 为基础[20,21],结合NTSYS 软件进行了桤木群体遗传参数估算与分析。
3.2 桤木Genomic-SSR 与EST-SSR 差异比较
本研究对两种来源SSR 标记的遗传差异进行了比较分析,结果显示:平均等位基因数与有效等位基因数EST-SSR 标记高于Genomic-SSR 标记,而两种杂合度Genomic-SSR 位点高于EST-SSR 位点,与前人研究结果均不完全一致。如在宋跃朋等与刘果等分别对杨树与桉树的研究中,均显示Genomic-SSR 标记的等位基因数、多态性、杂合度高于ESTSSR 标记[30,31],而在张亚东等对杨树研究则显示EST-SSR 标记等位基因数、多态性、杂合度高于Genomic-SSR 标记[32],而大豆相关研究则显示Genomic-SSR 的等位基因数高于EST-SSR 标记,但是EST-SSR 标记的多态性稍高于Genomic-SSR[33]。在对桤木群体遗传多样性的解析中,Genomic-SSR与EST-SSR 分析结果也不一致。尽管理论上由于EST-SSR 引物来自高度保守的DNA 转录区,其揭示的多态性在理论上应低于基因组SSR 标记,但由于试验材料与参试标记的不同,其显示的遗传差异可能不同。因此,在物种遗传多样性的研究中,应利用不同来源的分子标记进行综合评价,以获得更加客观的结论。
在对桤木群体遗传关系的分析中,在大的类群上Genomic-SSR 与EST-SSR 标记相一致,而在进一步的小类群上则显示出差异,而且本研究中两种标记计算的综合相关系数与Genomic-SSR 标记更为相似,表明本研究中Genomic-SSR 能更准确地揭示桤木不同群体或个体间的遗传关系,与前述宋跃朋等、刘果等与张亚东等研究结果不一致,这3 项研究均显示EST-SSR 能更准确地揭示基因型之间的遗传关系[30-32]。这可能与研究所用材料有关,这3 项研究分析的均是杨树与桉树不同种间的遗传关系,而本研究材料为桤木的不同群体,由于EST-SSR 来自DNA 转录区,保守性强,对亲缘关系较近的基因型灵敏度不及基因组SSR,但在近邻的种间具有通用性,对不同种属系统演化研究、加密遗传连锁图谱、基因精细定位、标记功能研究等具有重要作用。
总之,本研究表明桤木Genomic-SSR 与EST-SSR标记在解析群体遗传多样性与遗传关系方面有一定差异,要得到更准确的结果,需要综合使用两种标记。
