基于SNP标记的辣木群体遗传分析
2023-11-11普天磊韩学琴罗会英邓红山邹枚伶金杰夏志强王文泉
普天磊 韩学琴 罗会英 邓红山 邹枚伶 金杰 夏志强 王文泉
关键词:辣木;SNP;杂合度;群体结构;遗传多样性
中图分类号:S792.99 文献标识码:A
辣木(Moringa oleifera Lam.)属于辣木科辣木属的多年生落叶乔木,又被称为鼓槌树,辣木原产于印度,是埃塞俄比亚、尼日利亚、菲律宾和苏丹的重要农作物,在非洲、美洲等热带亚热带地区均有分布,在我国的云南、海南、福建、广州等地均有种植[1]。辣木有13 个种,其叶片中约含有20%~30%的蛋白质,叶片、花、果实含有丰富的维生素A、维生素B、维生素C 和钙、镁等矿物质,种子含高油酸,可用作化妆品、烹饪和机械润滑油,种子榨油后的剩余物可用于净化污水、饲喂动物[2-4]。同时辣木含有丰富的皂苷、生物碱、黄酮、酚类等次生代谢产物,具有抗氧化、抗炎、细胞保护、神经保护、抗癌等药理作用[5-6]。
SNP 标记相比较于RFLP、SSR 等传统分子标记而言,可检测单个碱基的插入、缺失、转换和颠换,具有变异数量多,分布广,遗传稳定性高,检测快、通量高的优点[7]。基于SNP 标记进行的遗传分析在植物学领域应用较多,例如,高嵩等[8]利用SNP 芯片进行玉米遗传多样性、群体遗传结构和类群间遗传关系分析,选育并审定了玉米新品种;韩志刚等[9]基于SNP 标记对148 份马铃薯种质遗传多样性进行分析,认为马铃薯绝大部分栽培种遗传相似性高,遗传背景不够丰富。目前,国内并没有利用SNP 分子标记对辣木种群的遗传学进行分析的报道。AFSM 技术为简化基因组测序技术,该法分别利用EcoR I-Msp I 和EcoR I-Hpa II 两种酶对基因组DNA 进行双酶切,并在两端加上区分不同样本的标签和接头,样品混合后进行双端测序,测序后获得的SNP 标记数量多,比传统分子标记更好地代表全基因组的遗传信息,具有成本低、准确性和稳定性高、易于操作的优点[10]。
杂合度分析有助于深入了解辣木的遗传组成情况,确定繁育类型,合理规划育种,传统研究繁育类型的方法主要是基于对花器官的形态学分析,传粉媒介的观察以及温室杂交试验展开,主要通过表型性状进行评估,易受环境、气候、栽培措施等因素影响,不能准确地反映植物基因型[11-13]。辣木群体结构的研究对于辣木种质资源的挖掘、利用和保护具有重要的理论和实践意义,遗传多样性及群体分化分析是遗传学研究的核心内容,亲本的遗传关系很大程度决定子代种子的质量,亲本间存在差异的遗传信息会随着杂交或自交过程传递给子代,使之在单核苷酸水平上呈现出来。目前辣木的繁育类型颇受争议,还没有学者基于SNP 对辣木的繁育类型进行研究,国内辣木育种工作进展缓慢,没有自主产权的辣木品种,存在种子繁殖会发生性状分离及种子管理不规范等问题,造成辣木优良品种缺乏、品种混乱的现象[14],辣木亲本和子代群体的遗传分析对于确定辣木繁育类型、分析亲缘关系及选育优良品种具有重要的意义。
本研究以96 份辣木为研究材料,结合基因组AFSM 高通量测序技术,与参考基因组进行比对后,进行基因型分析、杂合度分析、群体结构、遗传多样性、群体分化及连锁不平衡分析,以揭示辣木亲本与子代间的遗传关系,为辣木繁育类型和种质亲缘关系提供理论指导,以及为发掘控制辣木种质优良性状的优异等位基因提供理论依据。
1 材料与方法
1.1 材料
选取来源于同一母本通过自然授粉得到的YMLM002 辣木种子94 粒,该种质是经过连续3a的跟踪观测筛选出的果用型优良单株材料,具有产量高、果型好、种子饱满的特点[15]。辣木种子先用清水浸泡10 h,软化种子硬壳,再用100 mg/L高锰酸钾溶液浸泡0.5 h 消毒,清水洗净后点播于穴盘中(红土∶蛭石=1∶1),适时补充水分保证湿润,待苗长至15 cm 左右,收集94 份子代样品、1 份母株和1 份扦插苗样品备用。
1.2 方法
1.2.1 辣木基因组DNA 提取及建库 采用CTAB 法提取辣木样品DNA,用Nano Drop ND-1000 对DNA 样品浓度进行检测,并调节样品浓度至100 ng/μL,置于–20 ℃保存。采用AFSM 技术进行建库,利用EcoR I-Msp I 和EcoR I-Hpa II两种酶对96 份辣木DNA 样品进行混合双酶切,再将酶切产物连接加上用于区分不同样品的接头标签,纯化后进行PCR 扩增,样品混合后再用高通量测序平台Illumina 进行双端测序,并计算GC含量和Q30 评估测序数据质量。
1.2.2 辣木群体基因型分析 利用Perl 脚本对原始测序数据进行过滤,使用Bowtie 软件将过滤数据比对到辣木参考基因組ASM980114v1,再使用VCFtools 和BCFtools 软件检测并统计SNP 和Indel 位点信息。
1.2.3 辣木基因杂合度分析 使用AWK 语言分析96 份样品的杂合位点,并计算个体内基因的杂合位点比率即为个体内杂合度;同时通过将子代数据分别与亲本进行比对,找出差异位点,统计差异位点概率即为子代与亲本比对杂合度,分别生成个体内杂合度及子代与亲本比对杂合度统计图。
1.2.4 群体结构分析、遗传多样性及群体分化分析 利用Plink 对变异位点进行过滤,过滤掉最小等位基因频率低于0.05 及基因型缺失率小于5%的位点,哈迪–温伯格检验显著性P>0.0001,保留高质量的变异位点,再使用ADMIXTURE 软件进行群体结构分析,将亚群数K 值范围设置为1~10,根据得到的交叉验证错误率(cross-validationerror, CV error)值选择合适的亚群数K 值,以个体占亚群的遗传成分系数确定个体归属的类群,用R 软件绘制群体遗传结构矩阵图。采用GCTA 软件对过滤得到的高质量文件进行主成分分析,并用R 软件绘图;采用VCFtools 软件计算辣木群体的遗传多样性指数(π)及群体分化指数(Fst)。
1.2.5 连锁不平衡分析 使用LDBlockShow 软件,计算不同标记距离下的D值,并生成单体型块图以展示位点间的连锁不平衡程度。
2 结果与分析
2.1 辣木群体基因型分析
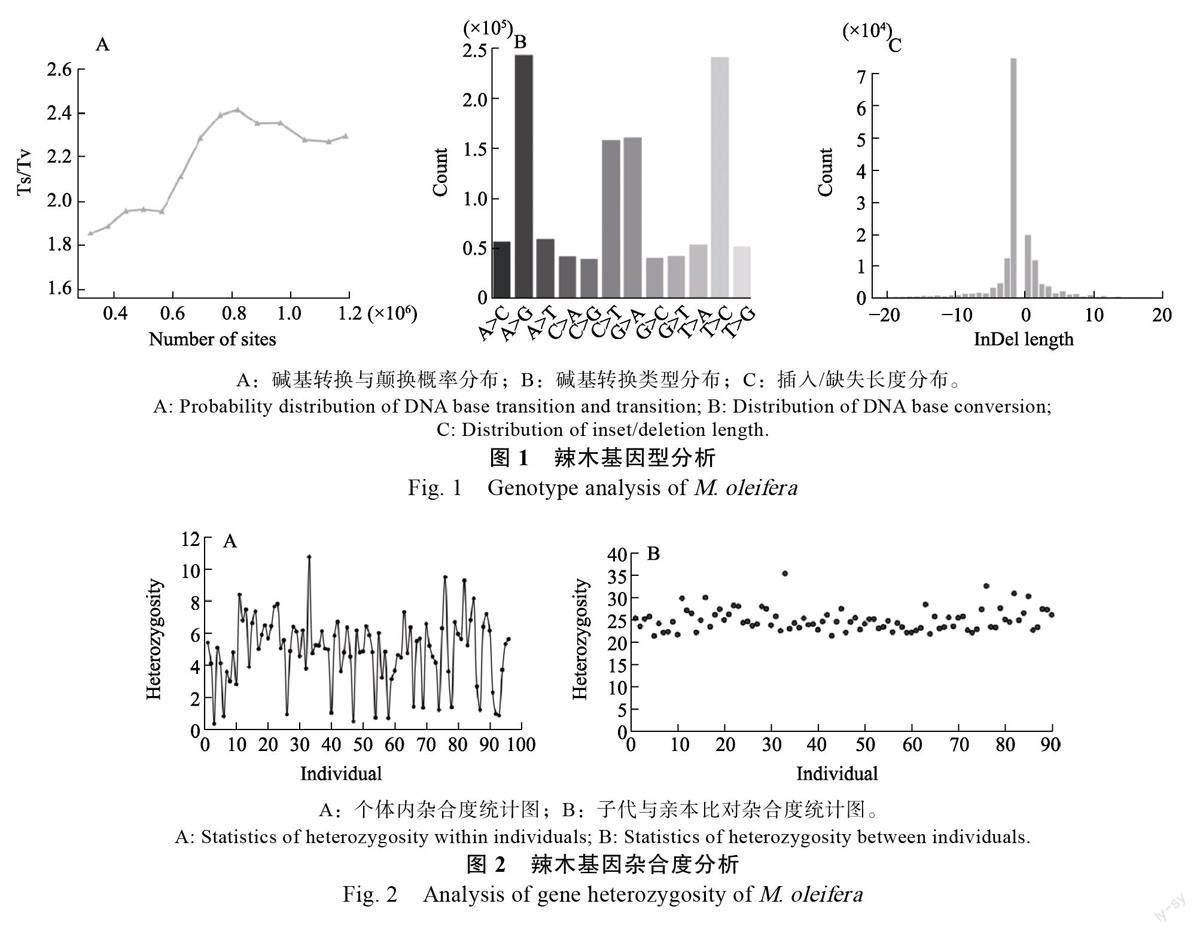
96 份辣木样品基因组DNA 经过AFSM技术建库、测序,将数据过滤并比对至辣木参考基因组ASM980114v1,参考基因组信息来源于NCBI 数据库(https://www.ncbi.nlm.nih.gov/data-hub/taxonomy/3735/),基因组大小为253.9 Mb,测序得到1.8 G 数据文件,346 615 757 条reads,测序长度为150 bp,平均GC 值为50.53%,平均Q30 为94.49%。采用VCFtools 和BCFtools 软件处理样品基因组数据后,得到1 187 831 个SNP 和150 861 个Indel位点,以及11 158 个多等位基因位点,4930 个多等位基因SNP 位点。SNP 同类型碱基之间的突变为转换,不同类型碱基之间的突变为颠换,SNPs发生转换概率与颠换概率的比值为2.08,单一序列发生转换次数为804 031,单一序列发生颠换次数为383 471(图1)。
SNPs 发生碱基转换和颠换概率随着位点的增大呈现先逐渐增加后缓慢降低的趋势(图1)。辣木不同类型的突变位点中,碱基转换的变异数量显著大于颠换的数量,其中碱基G/A 和C/T 的替换都较高,分别为243 672 和241 616 次;碱基A/G和T/C 的替换次之,分别为161 258 和158 648 次,碱基插入/缺失发生的次数随着碱基插入/缺失长度的增加而呈现出迅速下降的趋势。
2.2 辣木自然结实子代基因杂合度分析
采用1 187 831 个SNP 位点和150 861 个Indel位点分析96 份辣木样品的杂合度(图2)。辣木同源染色体上的SNP 位点为同一类型碱基,则该SNP 位点称为纯合SNP 位点,若为不同类型碱基,则为SNP 杂合位点。由图2A 可知,辣木个体内杂合度在10.79%~0.36%之间,个体内平均杂合度为4.89%,其中,母株杂合度为5.65%,扦插苗杂合度为5.34%。由图2B 可知,子代与亲本比对杂合度在21.22%~35.33%之间,子代与亲本的比对平均杂合度为24.85%。由此可知,导致辣木子代杂合的基因中,约有4.89%的基因为自身杂合基因,19.96%为外来遗传物质导致杂合的基因,基本表明辣木通過自花和异花2 种授粉方式繁衍后代。
2.3 辣木群体结构分析
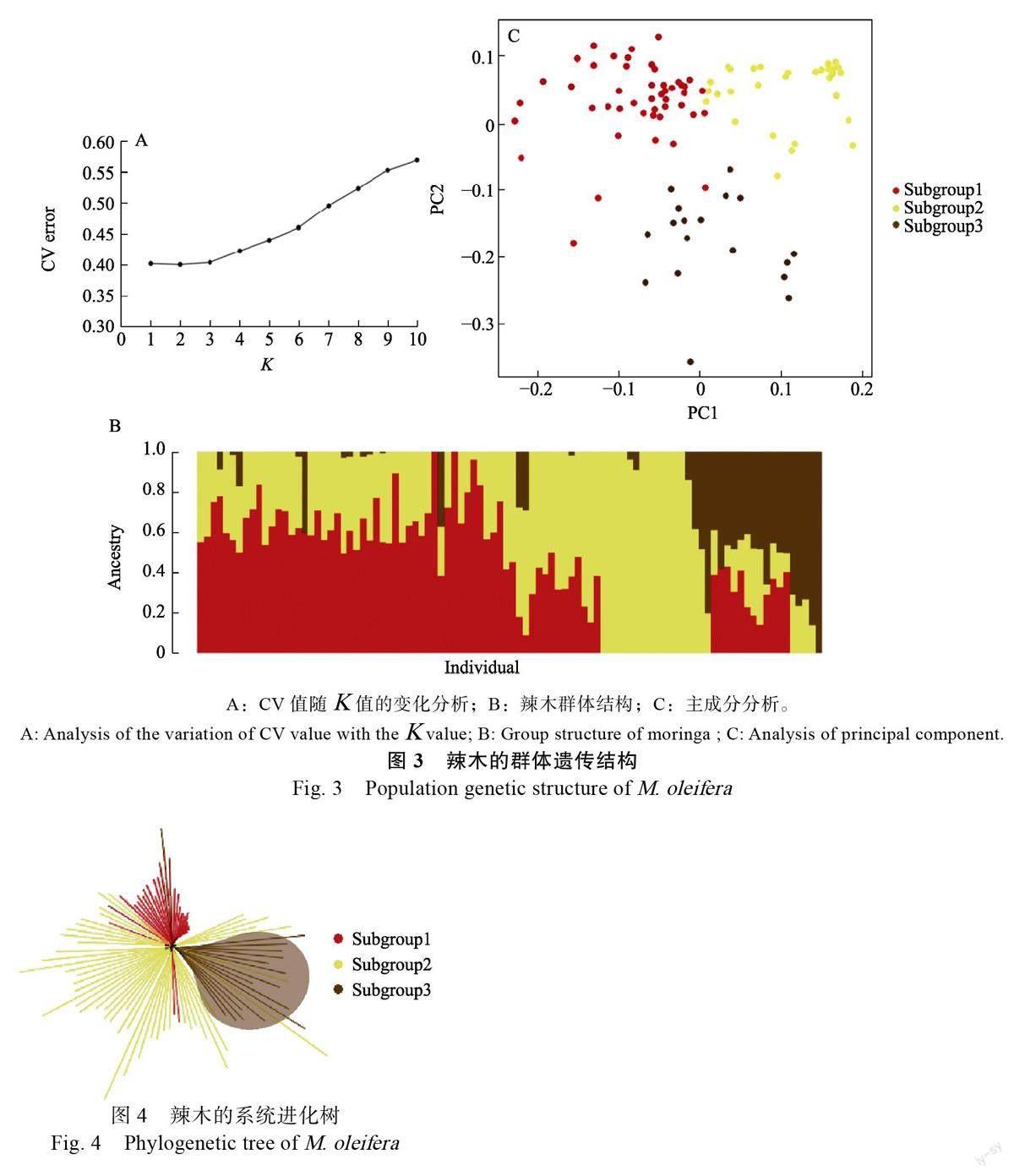
采用Plink 对变异位点进行过滤后,得到141 323 个SNP 位点,再通过软件利用所有SNP和Indels 分子标记对96 份辣木样品进行群体遗传结构分析,由图3A 可知,当K 值为3 时,随着K 值的增大,CV error 逐渐增大。由于K 值为2和3 时,CV error 值均较小且较为接近,分别为0.401 和0.404,但当K 值为2,即将96 份样品分为2 个亚群时,各亚群的个体呈现分布不集中的现象,故将96 份辣木样品分为3 个亚群(subgroup1-3)。根据个体在3 个亚群的Q 值,将个体归类到Q 值占比最大的亚群(图3B),发现3个亚群中分别有47、31、18 份材料,其中母株和扦插苗属于亚群1,亚群2、亚群3 均为子代样品。
主成分分析发现(图3C),亚群1 和亚群2在PC1 轴上有分布差距,而亚群3 与亚群1、2在PC2 轴上有一定的分布差距。大部分亚群可以聚类在一起,表明聚类结果与群体结构的划分具有一致性。同时,上述结果(辣木亲本与子代样本聚类为3 个亚群)再次论证了杂合度分析结果,即在生殖遗传的过程中,辣木并非以自花授粉的方式繁衍后代,在一定程度上接受了外来的花粉,导致后代在不包含母株的另外2 个群体中有分布。
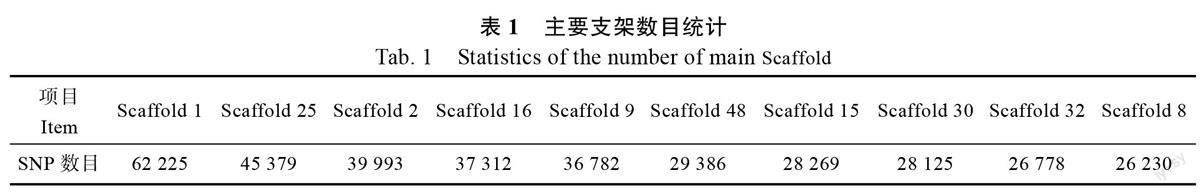
由图4 可知,群体进化树的聚类结果与群体结构的划分相一致,各亚群大致能聚在一起,且样品间有一定的交叉。相比较而言,亚群1 的分枝长度较短,有4 个个体分散在亚群2 中;亚群2 的分布总体集中,有7 个个体与亚群3 有交叉;同时,亚群3 有3 个个体与亚群1 有交叉。
2.4 辣木群体遗传多样性及分化分析
3 个亚群的π 值差距较小且均较低,平均π值也较低,为0.0010,表明96 个辣木群体的遗传多样性水平低。各亚群的Fst 在0.0049~0.0110 之间,其中亚群1 和亚群2 间的Fst 值最小,亚群2和亚群3 间的Fst 值最大,各亚群间的Fst 值均小于0.05,表明各样本之间存在较弱的遗传分化(当Fst 等于0 或1 时,分别表明亚群间没有分化或完全分化;当Fst 为0~0.05 时,表明亚群间的分化较弱;当Fst 为0.05~0.15 时,表明亚群间为中度分化;当Fst 为0.15~0.25 时,表明亚群间的分化较强[16]),各亚群间的亲缘关系相对较近。
2.5 连锁不平衡分析
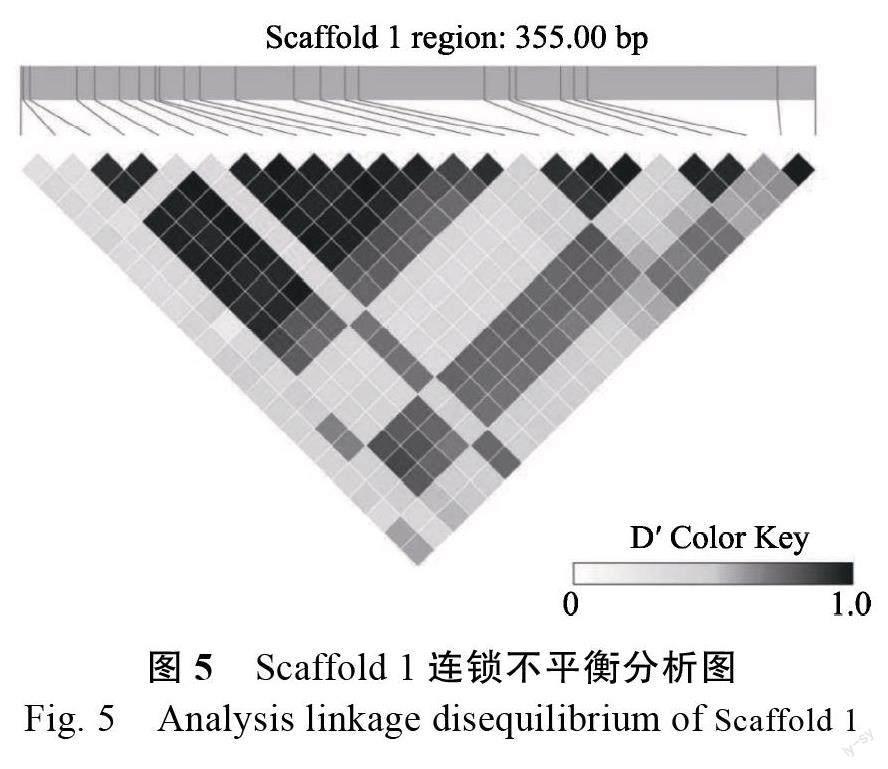
结合多态性核心SNP 位点在辣木基因组上对应位置分析,发现共有136 个Scaffold,主要Scaffold 统计情况见表1(SNP 数量前十的Scaffold)。其中,Scaffold 1 的SNP 数目最多,为62 225个,Scaffold 122 的SNP 数目最少,为288 个。通过LDBlockShow 软件对Scaffold 1 在6.748~6.749 Mb 区域内的变异信息进行连锁不平衡分析,发现6 748 044~6 748 185 位点之间具有强连锁不平衡关系,而6 748 040 与6 748 041、6 748 041与6 748 044 等位点间的连锁关系弱(图5)。
3 讨论
AFSM 技术采用EcoR I-Hpa II 和EcoR I-MspI 两组双酶切体系简化基因组DNA 的复杂度,目前已发展得较为成熟,已用于检测巴西木薯、澳洲坚果、麻疯树等植物的SNP、Indel 及甲基化位点[17-19],该技术DNA 处理步骤和数据分析步骤相对简单,效率高,测定的位点稳定,无需进行超声剪切或荧光标记,试验成本低,适用于大量非模型物种的基因分型。本研究利用该技术得到1 187 831 个SNP 和150 861 个Indel 位点,可实现辣木亲本及子代遗传分析的目的。
国内外相关学者从不同的角度对辣木的繁育系统进行研究,吕亚等[20]发现狭瓣辣木在開花第一天就有花粉活力和微弱的柱头可授性,且开花之初柱头高于雄蕊,之后逐渐低于雄蕊。MULUVI等[21]利用AFLP 分子标记研究肯尼亚种源辣木的繁育系统,表明该种源辣木种子是自交和异交的混合产物。起国海[22]研究辣木对干热河谷传粉网络的影响,并表明辣木单花能提供5~30 μL,含糖量高达60.5%的花蜜报酬物,属于昆虫传粉植物,主要传粉者为蜂类。本研究中辣木个体内平均杂合度4.89%,子代与亲本的比对平均杂合度为24.85%,表明辣木繁殖方式为自交与异交同时存在。因此,在进行辣木杂种优势利用时,需要关注相关个体间的随机化分布和最小距离,以最大限度地增加差异品种/系间的杂交受精,并尽量减少品种内部的自交。
植物的繁育系统、选择、遗传漂移、突变和迁移是影响植物群体遗传结构的进化因子[23],本研究利用ADMIXTURE 软件对辣木的群体结构进行分析,将96 个辣木群体划分成3 个亚群,该结果与聚类分析和主成分分析的结果类似,3 种群体结构分析方法相互补充印证,表明辣木群体的遗传结构划分可靠。在群体结构划分中,大部分亚群可以聚类在一起,其中,1 亚群有亲本及子代样品,而2、3 亚群均为子代样品,该结果表明辣木自然结实子代群体除了携带亲本的遗传信息外,还携带有外来的遗传信息,即辣木繁衍后代的方式不仅为自花授粉,而且还存在异花授粉。子代样品在3 个亚群中均有分布,可能是由于亲本植株种植于保存有不同辣木种质的资源圃内,不同来源材料的花粉传播至亲本植株所导致的。
群体的π 值和Fst 值是衡量群体遗传分化程度的重要参数,RAJALAKSHMI 等[24]使用ISSR、SRAP 标记研究印度种源辣木的遗传多样性,表明辣木的平均遗传分化系数为0.15,总遗传多样性指数为0.17。本研究发现辣木群体的π 值为0.0010,Fst 值在0.0049~0.0110 之间,表明本研究所用辣木群体遗传分化弱,遗传多样性较低,该现象应该是由于选取的辣木群体是亲本与子代亲缘关系较近造成的。同时,也表明引起子代杂合的外来基因与亲本的基因型差异不大,这可能是由于本研究文采用的是栽培种辣木资源,经历了多次的人工选择育种,资源间丰度低造成的,后续可引进印度、非洲种源的优良辣木种质,以丰富资源圃内的辣木种质[25]。
当2 个距离较近的等位基因在同一单体型上同时出现的频率高于随机出现的频率时,表明它们处于连锁不平衡状态。在定位克隆中,通过连锁可检测到产生连锁信号的变异,在关联分析中,利用邻近位点形成的强连锁不平衡,有助于找到与性状相关的位点[26-27]。本文对辣木的SNP 位点进行了连锁不平衡分析,并在单体型块图上发现了连锁不平衡关系强的基因区域,可为研究多个处于连锁不平衡的位点与重要性状的关联性提供参考依据。
