基于真实世界数据的随机对照试验模拟研究设计、评价与实例分析
2023-11-10刘佐相
刘佐相
北京大学公共卫生学院流行病与卫生统计学系
重大疾病流行病学教育部重点实验室(北京大学)
龙子临
北京大学公共卫生学院流行病与卫生统计学系
重大疾病流行病学教育部重点实验室(北京大学)
赵厚宇
北京大学公共卫生学院流行病与卫生统计学系
重大疾病流行病学教育部重点实验室(北京大学)
詹思延
北京大学公共卫生学院流行病与卫生统计学系
重大疾病流行病学教育部重点实验室(北京大学)
北京大学第三医院临床流行病学研究中心
北京大学人工智能研究院智慧公众健康研究中心
宋海波
国家药品监督管理局药品评价中心
国家药品监督管理局药物警戒研究与评价重点实验室
孙凤*
北京大学公共卫生学院流行病与卫生统计学系
重大疾病流行病学教育部重点实验室(北京大学)
对上市后药品开展真实世界研究(real world study,RWS)可以获得相应的真实世界证据(real world evidence,RWE),以帮助临床医生或监管部门进行相应的决策。但是目前开展的RWS 还未能在卫生技术评估方面充分发挥应有的作用,这与其尚有诸多局限性有关,如研究类型与研究目的不符、设计方案与研究类型不符、研究对象的选择不严谨、暴露或干预定义不明确、组间可比性差、观察指标选择不规范、缺少数据质量控制和统计学校正,因此可能无法很好地回答某一具体临床问题[1]。
随机对照试验(randomized controlled trial,RCT)是最高级别的医学证据,这是因为其研究设计往往具有以下优点:①明确的纳排标准,帮助了解结果的外推性;②详细的干预措施定义,减少信息偏倚;③随机化分组,保证组间基线的可比性,控制混杂;④选取的结局常用生理学或生物医学指标来描述,避免主观性和测量偏倚,且有利于解释生物学基础反应[2]。此外,RCT 往往还采用盲法,进一步避免了研究过程中可能产生的选择偏倚和信息偏倚。但是,RCT 研究通常周期较长,花费大量人力、物力,仅依靠RCT 进行干预措施效果评价不利于医学技术的发展,因此需要改进和规范RWS 以推动高质量医学证据的产生。为此,有学者提出了仿照RCT 开展观察性研究的思路,既借鉴了RCT设计优势,增强了因果推断的强度,又保留了RWS 的代表性,可以作为打通药品上市前与上市后证据链的桥梁,以期为医疗决策提供合理的证据支撑。
1 研究设计思路
任何医学研究设计都必须基于某一具体的临床问题,即在哪类人群中,哪种干预措施相较于哪种对照措施,在哪个结局指标方面有怎样的效果,可简化为研究对象(Population,P)、干预措 施(Intervention,I)、对 照措施(Comparison,C)、结局(Outcome,O)四部分。
有研究者认为,回答干预措施效果的最优研究设计应是具有临床相关结果(即无替代终点)、长期随访和完美依从性的大型随机试验,次优选择便是使用真实世界数据(real world data,RWD)模拟目标试验(emulate a target trial)的观察性研究[3],目前也有译为RCT 模拟研究、RCT 仿真研究等。研究步骤分为两步:先基于临床问题构建一个目标试验,该试验可以是已注册或发表的RCT,也可以是经过专家讨论形成的理论上的RCT,参照目标试验关键特征构建相应的RWS 方案;再使用RWD 按照既定研究方案进行数据分析,最终得到相应结果。
2 随机对照试验模拟研究的方案设计框架及要素
一项高质量的RCT 研究方案通常包括试验设计类型、纳排标准、干预措施定义、结局指标定义、样本量计算、随机方法、盲法、因果对比策略和统计分析方法。RCT 模拟研究方案应参照RCT 研究方案并根据RWS 特点进行相应调整。除了目前已开展的RCT 模拟研究仅考虑平行对照设计,以及使用与RCT 相同的样本量计算参数,根据目标试验研究方案制定相应的RCT模拟研究方案,主要考虑以下7个关键特征:纳排标准、治疗策略、分配程序、随访、结局定义、因果对比和统计分析策略。其中RWD 本身是在开放状态下被收集的,无法模拟盲法和随机分配,因此分配程序的模拟主要通过使用相应的统计分析方法以减少和控制相应的偏倚。
2.1 纳排标准
当某些人群基线特征对干预措施存在效应修饰作用时,同一干预措施在不同特征的人群中,会表现出不同的效果。例如有研究发现,基于相同源人群开展的糖尿病相关RCT,与具有高度代表性的糖尿病登记研究相比,RCT 参与者的平均年龄更小,女性参与者的比例也更低[4]。人群特征主要是由纳排标准决定的,因此在开展RCT 模拟研究时,应尽可能采用与目标试验相同的纳排标准。同时,纳排标准的制定还需要考虑观察性研究中容易产生的相关偏倚,如若不对研究对象治疗开始后进入队列的时间进行限制,可能会出现由于现用药者是治疗早期阶段的幸存者,而排除了因用药早期事件而停止用药的患者,导致现用药者偏倚。新用药者队列设计可以在极大程度上避免这一问题,即仅纳入一定未治疗时间(洗脱期)后接受特定治疗的患者。此外,还要注意纳排标准不能包括对基线后事件的限制,否则也会造成偏倚[5]。
2.2 治疗策略、随访及结局定义
治疗策略、随访及结局定义是决定研究间的临床异质性的重要部分。若定义标准存在差异,则反映为研究结果的统计学异质性。因此,RCT 模拟研究相关定义必须尽可能靠近目标试验,减少研究间的临床异质性。但是,由于观察性研究中的干预测量、结局测量全部来自于现有的真实世界数据,数据的质量难以被把控,且无法捕捉全部信息,可能会存在信息偏倚。因此,在制定RCT 模拟研究方案时,还需要对可能存在的模拟差异和偏倚进行控制。
RCT 往往对干预措施具有明确的定义,包括药品种类、剂型、剂量、给药方式等,而在真实世界中很难找到采用与RCT 完全相同的治疗模式的患者,此时应当考虑暴露与结局间的生物学作用机制,制定合适的干预措施定义标准[6]。
对照组的选择也十分重要,不同的选择可能会导致不同的偏倚[6]。需要注意的是,RCT 中的安慰剂对照很难在真实世界中进行模拟。如果直接使用空白对照代替安慰剂对照,干预组患者与对照组患者在疾病严重程度与不良事件风险方面将具有较大差异,可能导致混杂偏倚。对此,有相关文献仍推荐在评估干预措施的有效性与安全性时,使用阳性对照可能更稳妥[6]。
在确定随访开始时点时,容易产生时间相关偏倚。这是因为观察性研究中每个个体满足纳排标准的时间和干预给予时间往往是不同的,处方时点往往会晚于相关诊断时点,两组随访起点不一致而使得随访起点到干预开始之间的时间被剔除或错分为暴露人群时,就会导致永恒时间偏倚[7],因此需要谨慎考虑入组时点、暴露时点以及观察起始时点三者的界定。可以仅研究能度过永恒时间的患者,即把随访开始时间点设置为永恒时间的结束点(干预给予时点),但此方法仅适用于阳性对照。对此,有研究者在模拟目标试验方法框架中提出“模拟-删失-加权”方法[8],即每一个入组患者(真实患者)都对应着一个克隆患者,双方被分配到不同的干预组,在指定的宽限期内,当真实患者接受的干预措施偏离既定分组干预措施时,则将克隆患者做删失处理,若在宽限期内发生死亡等删失事件,则对两个患者都做删失处理。然而,由于这种删失是不随机的,可能会导致选择偏倚,这种潜在偏倚可以在分析中使用逆删失概率权重予以控制。
2.3 因果对比与统计分析方案
RCT 中常用的因果对比策略包括意向治疗(intention-totreatment,ITT)分析和符合方案(per-protocol,PP)分 析,RWS 中应采用与RCT 相同的因果分析策略。RCT 中由于随机分组且患者依从性较好,通常使用ITT 分析作为主要分析策略。但是由于RWS 中无法通过随机化而保证组间基线可比性,因此,通常需要采用匹配、分层、标准化、多因素回归、协方差、断点回归等方法校正混杂因素的影响[9-11]。但是当需要校正的混杂因素较多时,采用以上方式将不再合适。此时,基于倾向性评分的匹配、分层、加权、回归等方式可能更加合适[12]。但是,当包含过多仅与暴露相关的协变量或无关的变量时,可能会使方差膨胀而不能降低偏倚,此时使用高维倾向性评分可能是一个不错的选择[13]。但传统的倾向性评分只能校正基线混杂,当暴露和混杂具有时变性时,其不再适用。因此,当要模拟PP 分析策略时,有必要使用G 方法对时依性变量进行调整,以校正治疗依从性。其中,基于逆概率加权的边际结构模型已被应用到RCT 模拟研究之中[14]。
但是G 方法的使用前提是假定不存在未测量混杂,对于未观测混杂的潜在影响,目前相关方法尚不成熟,寻找合适的工具变量进行定量评估较为困难,但可以通过E 值(E value)[15]和阴性对照[16]等方法进行校正或定性评估剩余混杂。
除了上述控制混杂的相关统计方法外,RCT 模拟研究还应该使用与目标试验相同的统计学描述和统计推断方法,包括置信区间估计及假设检验。此外,还要详细描述除主分析以外的亚组分析和敏感性分析策略。亚组因素方面,需要考虑可能导致效果异质性的因素(如遗传学、人口学、病理生理学、环境、合并症、合并用药、居住区域、保险类型等),帮助进行更加精细化的临床决策。此外,还要考虑在数据分析过程中采取的不同方法,如针对缺失数据的不同处理机制、不同的协变量组合等,对其进行敏感性分析,从而探讨结果的稳健程度[17-18]。
2.4 初步可行性分析及研究方案注册
制定研究方案后,应对其进行初步的可行性分析。这是因为应用纳排标准进行人群筛选以及使用倾向性评分校正基线混杂后,可能有大量个体被排除,从而影响结果的稳健性和外推性。应用纳排标准和简单的混杂校正,判断RCT 模拟研究最终纳入的研究对象数量能否满足统计效能。若样本量过低,则应当终止研究。
若初步可行性分析结果支持该RCT 模拟研究的开展,为了避免研究结果的选择性报告,增加研究透明性,应在研究开始前在相关网站(如ClinicalTrials.gov)进行注册和研究方案公布。
3 随机对照试验模拟研究方案设计实例解读
美国食品药品监督管理局(FDA)为了加速RWS 进展,资助了一系列RWS 计划,其中包括由哈佛大学医学院附属布莱根妇女医院(Brigham and Women's Hospital)以及一家医疗保健技术公司(Aetion)共同牵头发起的RCT Duplicate项目。该项目旨在利用3 个医疗保险索赔数据库,使用RCT 模拟研究方法模拟30 个已发表的RCT,以推进RWS 进展,并探讨使用RCT 模拟研究方法重现RCT 结果的能力,以及分析模拟差异的原因[19]。
PLATO 试验是一项评估氯吡格雷75mg/ODD(每日一次)与替格瑞洛90mg/BD(每日两次)心血管保护作用差异的临床试验(注册号为NCT00391872)[20],相应的RCT Duplicate 模拟研究[21](注册号为NCT04237935)已 在ClinicalTrials.gov 网站上注册并描述了详细的研究计划和统计分析方案,笔者对该RCT 模拟研究方案设计进行解读如下。
3.1 设计流程图
设计流程图有助于直观理解RCT 模拟研究设计(图1)。在PLATO 模拟研究中,首先将既往具有急性冠状动脉综合征入院记录且之后具有氯吡格雷或替格瑞洛处方记录的患者纳入队列,并将处方时间定为指示日期。将指示日期前180 天设置为洗脱期,以选择新用药者而避免现用药者偏倚。纳排标准仅包含对指示日期前相关条件的限制,以避免对基线后事件的限制而产生的选择偏倚。对指示日期前180 天内的相关协变量进行测量,以用于倾向性评分匹配。从指示日期后的第1 天开始对患者进行随访,直到所有删失事件发生的最早时间作为随访结束日期。
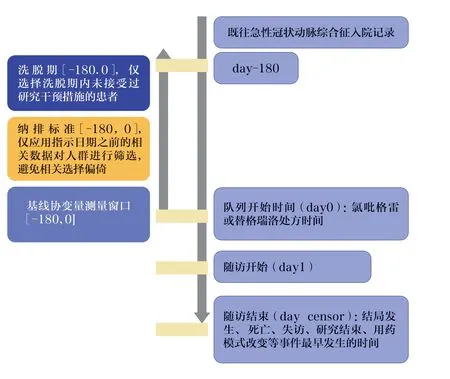
图1 PLATO 试验模拟研究设计流程图
3.2 纳排标准
RCT 模拟研究要尽可能应用与RCT 相同的纳排标准。PLATO 模拟研究方案中对于每一条RCT 纳排标准都进行了模拟,给出了相应的定义,并根据每一条纳排标准存在模拟差异的程度(如充分模拟、较好模拟、较差模拟、无法模拟但对结果影响不大),用不同颜色加以标注。例如,RCT 纳排标准中对于急性冠状动脉综合征入院的患者定义为:由于心电图的ST 段抬高或非ST 段抬高急性冠状动脉综合征入院,且在入院前24 小时在静息状态下发生过由于动脉粥样硬化而导致的发作时间超过10 分钟的心肌缺血的患者。RCT 模拟研究中对该条标准的定义为:在入院日期前的两个星期内具有急性心梗相关诊断(ICD-9:Dx 410.X 排除410.x2;ICD-10:I21.0x,I21.1x,I21.2x,I21.3x,I21.4x,I22.0x,I22.0x,I22.1x,I22.2x,I22.8x,I22.9x,I25.2x)以及不稳定性相关诊断(ICD-9:411.x ICD-10: I24.1x,I20.0x,I25.1x,I25.7x,I24.0x,I24.8x,I24.9x),并将此条标准的模拟差异判断为较好模拟。而对于RCT中年龄≥18 岁的限制,则可以使用入院病历中的年龄数据进行充分模拟。但是由于数据库中缺乏实验室指标检测数据,无法模拟RCT 纳入标准中高于正常水平的肌钙蛋白Ⅰ、肌钙蛋白T 或肌酸激酶同工酶(CK-MB)等提示心肌坏死的阳性生物标志物,可能对研究结果造成较大影响。此外,PLATO 模拟研究方案还提供了人群纳排流程表,进一步展示人群纳排过程,帮助人们了解真实世界人群特征。
3.3 干预措施、结局和随访定义
PLATO 模拟研究尽可能模拟了RCT 中的干预措施与结局定义。由于很难从数据库中寻找相同剂量的药品使用者,在RCT 模拟研究中只对药品的种类进行模拟,而对RCT 中的心肌梗死、脑卒中和死亡结局的模拟给出了具体的国际疾病分类(ICD)编码。
PLATO 模拟研究方案中明确指出了随访是从干预措施最早处方记录的第2 天开始,直到最早的终点事件发生结束。终点事件包括结局发生、退出数据库、用药改变以及研究终止等。
3.4 因果对比与统计分析策略
PLATO 模拟研究方案中指明分别使用ITT 和As-treated(AT)两种分析策略,考虑到RWS 中患者依从性较差,将AT 分析策略作为主分析策略。PLATO 模拟研究方案中指明使用1 ∶1 倾向性评分匹配的方法来校正混杂因素,且指定了需要纳入倾向性评分的相关变量,并进行了基线描述。其中,并非所有的数据库都包含患者的实验室检查数据,因此实验室检查指标并不被纳入最终的倾向性评分模型。并且,考虑使用不对称修剪的倾向性评分匹配方法,去除干预组中倾向性评分低于2.5 百分位数和对照组中倾向性评分高于97.5 百分位数的患者再进行匹配,作为敏感性分析。使用Cox回归模型,进行效应值估计。
4 小结

药品等医疗干预措施必须通过医疗卫生监督管理部门的卫生技术评估,确保其在人群中应用的获益大于风险,才能在患者中进行推广。目前的卫生技术评估依据主要为上市前的RCT 证据,这是因为RCT 能够最大可能保证结果估计的无偏性,确保结果的内部有效性。但是,RCT 也具有一些局限性,包括:对于罕见病和严重疾病,由于难以选择对照组或者涉及伦理问题,仅能依靠单臂研究获取证据;在开展RCT 时,为了评估干预措施的疗效,多数研究选择安慰剂对照,而缺少头对头阳性对照以帮助更好地进行临床决策;RCT 纳排标准严格,样本量受限,可能无法估计干预措施在更多患者亚群中的效果;RCT 通常开展时间较短,难以评估远期和罕见不良事件。因此,必须开展针对干预措施的上市后再评价,以提供RCT无法提供的真实世界有效性和安全性证据。
RCT 模拟研究综合考虑了以上场景,为卫生技术评估提供了合适的证据支撑,也获得了美国FDA 等机构的关注,且越来越多的研究者开始采用RCT 模拟研究的方法开展RWS,药品和医疗器械的上市后再评价就是基于RWD 的RCT 模拟研究的重要应用。RCT 模拟研究是以RCT 设计为蓝本,对观察性RWS 方法加以改良,在保留RWS 结果较好代表性的基础上,提高结果估计的准确度,以获得更高质量的医学证据。通过对RCT 研究方案中纳排标准、干预措施、结局定义以及随访观察的模拟,使用合适的混杂校正方法,获得高质量RWE,从而具有回答在上市前阶段RCT 无法回答的临床问题的潜力[22]。
尽管RCT 模拟研究具有以上优势,但其局限性仍需要被重视。需要注意的是,RCT 模拟研究依靠常规收集的医疗数据,数据收集过程缺乏明确的研究目的,因此可能会缺少部分变量,影响RCT 模拟研究方案的施行,从而产生模拟差异。因此,在开展RCT 模拟研究之前,必须要评估RWD 的可获得性、适用性与质量,以及明确研究设计与统计分析方法。当使用单一数据源缺乏某些关键变量时,可以考虑使用多数据源,确保数据源能够涵盖模拟过程所需要的核心变量。
此外,RCT 模拟研究本质上仍然是观察性研究,旨在为开展基于RWD 的观察性研究的设计与分析提供指导性框架,因此仍然无法完全避免观察性研究本身存在的问题(如测量偏倚和混杂偏倚),需要慎重评估证据质量。例如,RCT 模拟研究无法采用随机化,传统的倾向性评分方法仅能校正部分基线混杂,对于未测量的混杂缺乏控制手段;同时,已开展的RCT 模拟研究很少考虑混杂因素的时变性,会对结果的估计产生影响。因此,进一步发展控制混杂的检测方法,如工具变量法、G 方法等在RCT 模拟研究中的应用,对于提高RCT模拟研究质量具有重要意义。为了便于将研究结果用于卫生技术评估决策,在撰写报告时应遵循观察性研究报告规范,如加强观察性流行病学研究报告的声明(strengthening the reporting of observational studies in epidemiology,STROBE)[23]、使用常规收集卫生数据开展观察性研究的报告规范(the reporting of studies conducted using observational routinelyc ollected health data,RECORD)[24]等,以保证研究报告的完整、透明和清晰,并在应用证据时使用ROBINS-I(risk of bias in non-randomized studies of interventions)[25]等非随机干预研究偏倚风险评估工具进行研究质量评估。
