不同产地赤芝药材的质量分析
2023-11-08谢孟君祝子喻俞月婷
曾 秒,谢孟君,祝子喻,俞月婷,张 梅
不同产地赤芝药材的质量分析
曾 秒,谢孟君,祝子喻,俞月婷,张 梅*
成都中医药大学药学院 西南特色中药资源国家重点实验室,四川 成都 611137
对不同产地批次赤芝进行全面的质量评价,为灵芝药材质量标准的完善及优质赤芝原料药的筛选提供依据。收集15批次各GAP基地的灵芝药材,参照《中国药典》2020年版方法及行业通用方法,考察其水分、灰分、水溶性浸出物、醇溶性浸出物,分别采用比色法、柱前衍生高效液相色谱法(PMP-HPLC)、高效凝胶渗透色谱法(HPGPC)测定多糖含量并进行比较;采用比色法测定三萜甾醇含量,HPLC法进行灵芝酸A一测多评;同时对15批赤芝的HPLC指纹图谱进行差异比较。15批赤芝药材均符合《中国药典》2020年版的质量标准要求,指纹图谱聚类分析和主成分分析(principal component analysis,PCA)结果表明山东聊城产赤芝能明显区别于其他批次样品。15批赤芝药材均符合《中国药典》质量标准,说明国内赤芝GAP基地种植质量良好;同时山东聊城产赤芝能显著区别于其他产地批次赤芝样品,主要体现在三萜甾醇等有效成分的含量更高。
灵芝;质量评价;主成分分析;多糖;三萜;指纹图谱
灵芝是多孔菌科真菌灵芝的子实体,由于极高的药用价值而被大规模商业化种植,其中赤芝(Leyss. ex Fr.) Karst.和紫芝Zhao Xu et Zhang被纳入《中国药典》2020年版[1]。古代认为灵芝具有死而复生、延年益寿的功效,中医用于心神不定、不茶不饭、失眠梦多、肺气虚证咳喘等。现代研究表明,灵芝在肝损伤[2]、癌症[3-4]、心血管疾病[5]、大脑损伤[6]、氧化损伤[7-8]、高血糖[9-10]、炎症[8,11]、免疫[12]、衰老[13]等疾病上面发挥了巨大作用,有非常重要的研究价值和市场前景。目前灵芝在食品药品方面已经有广泛的应用[14],我国的灵芝GAP基地有必要按照更严格的质量标准,逐步规范化和标准化,不断推动灵芝产业的发展。
灵芝的主要化学成分为多糖类、三萜类化合物、甾醇、氨基酸类、生物碱、核苷类、脂肪酸类以及微量元素等[15-16],其中多糖和三萜是其主要活性成分,也是研究最多的成分。经过多年的培育,灵芝形成了诸多品种,形态、质地各异,成分含量也有所区别,有效成分质量控制的方法也多种多样[17-18],其中赤芝在我国应用广泛,在各个省市都有扩大栽培的趋势,但赤芝培育的品种繁多,同时GAP基地的种植水平不一。
本实验选用15批不同GAP种植基地的赤芝药材,对其进行全面的质量分析,为不同GAP基地赤芝的种植现状提供质量依据。
1 材料
1.1 药材来源
分别从5个省份的11个灵芝GAP种植基地收集15批赤芝药材,由广东省微生物研究所食用菌中心谢意珍研究员鉴定为多孔菌科真菌赤芝(Leyss. ex Fr.) Karst.的干燥子实体。药材样品来源见表1。
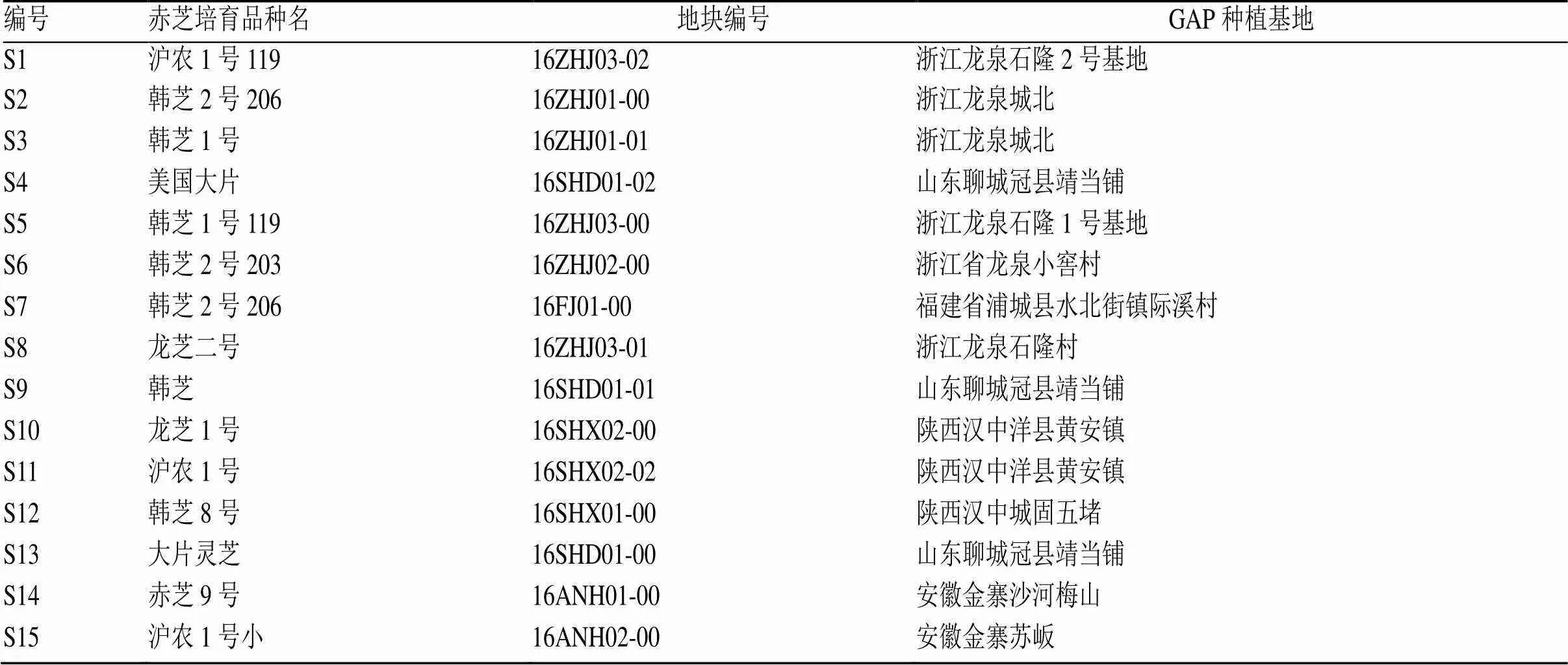
表1 灵芝供试样品编号、各培育品种及GAP种植基地
1.2 试剂
对照品齐墩果酸(质量分数为98%,批号MUST-16070406,)购于中国科学院成都生物研究所;葡萄糖(质量分数为99.5%,批号11083-201609)、岩藻糖(质量分数为99.7%,批号112014-201601)购于中国食品药品检定研究院;来苏糖(质量分数为99%,批号L115557)、甘露糖(质量分数为98%,批号D121716)、葡萄糖醛酸(质量分数为98%,批号C10584817)、半乳糖(质量分数为97%,批号G100368)购于上海阿拉丁试剂有限公司;右旋糖酐相对分子质量标准(套)对照品(批号140637~646-201203)购于Sigma公司;灵芝酸A(质量分数为99%,批号A0842)、灵芝酸F(质量分数为97.3%,批号A0917)、灵芝酸H(质量分数为97.2%,批号A0931)、灵芝酸B(质量分数为99%,批号A0916)、灵芝烯酸B(质量分数为99%,批号A0923)、灵芝酸C2(质量分数为99%,批号A0911)、灵芝烯酸C(质量分数为99%,批号A0924)、灵芝烯酸D(质量分数为98.7%,批号A0907)、灵芝酸D(质量分数为99%,批号A0906),灵芝酸G(质量分数为99%,批号A0909)均购于上海诗丹德标准技术服务公司;乙腈为色谱级;三氟乙酸、甲醇、乙醇、浓硫酸、蒽酮、冰醋酸、高氯酸、醋酸乙酯、香草醛、叠氮化钠、硫酸钠、盐酸、氢氧化钠、醋酸铵等均为分析级。
1.3 仪器
GZX-9070MBE型电热恒温鼓风干燥箱(上海一恒科学仪器有限公司);SX-2-5-12型箱式电阻炉(北京科伟永兴仪器有限公司);UV3200S型紫外分光光度计仪(上海美普达仪器有限公司);安捷伦1200高效液相色谱仪(美国Agilent公司)、示差折光检测器(美国Agilent公司)。
2 方法与结果
2.1 一般性检查及浸出物
按照《中国药典》2020年版四部通则0832项下的烘干法和2302项下的灰分测定[19],检测15批样品的水分及灰分,结果见表2。15批样品均满足《中国药典》的要求,水分为1.47%~7.28%,灰分在0.69%~2.79%。
因灵芝水溶性浸出物得率太低,同时为方便同步进行多糖相对分子质量分布、多糖含量、单糖组成测定,水溶性浸出物参考《中国药典》2020年版设计方案[1]。取赤芝粉末(过2号筛)8 g,精密称定,置500 mL圆底烧瓶中,精密加水240 mL,静置1 h,加沸石2~4颗,置电热套中,连接回流装置,加热至沸腾,并保持微沸4 h,趁热抽滤,用25 mL热水润洗3次,往滤渣及滤纸中再精密加水240 mL,加热回流3 h,操作同上,合并滤液。加水至640 mL,摇匀,量取320 mL(约为4 g样品),置已恒定质量的蒸发皿中,水浴蒸干后,于105 ℃干燥3 h,置干燥器中冷却30 min,迅速精密称定干燥物质量,结果见表2。15批样品的水溶性浸出物含量符合《中国药典》2020年版的要求,质量分数为6.50%~16.43%。
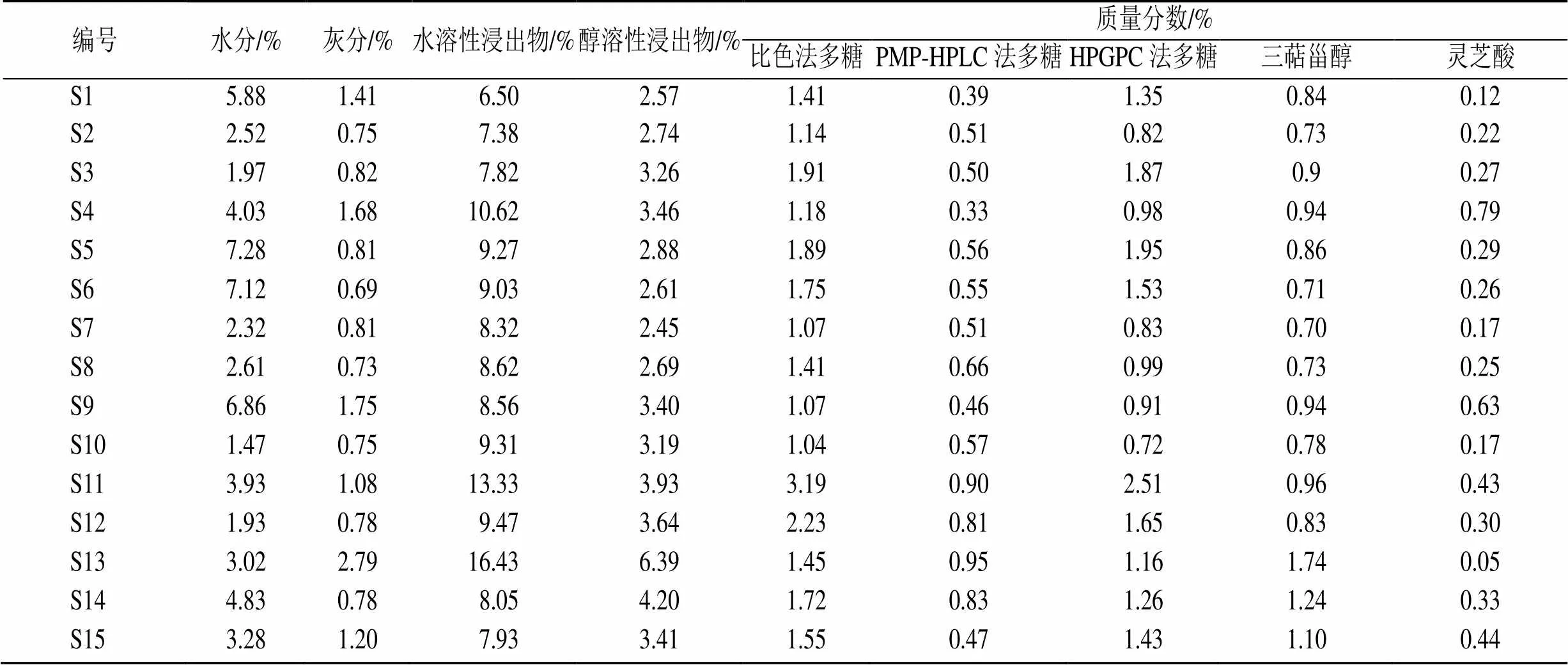
表2 赤芝质量评价结果
灵芝醇溶性浸出物参照《中国药典》2020年版通则2201[19],以水溶性浸出物的方法,将溶剂替换为乙醇,设计步骤如下:灵芝子实体打粉,过筛,取2~5号筛之间的粉末约4 g ,精密称定,置250mL平底烧瓶中,精密加入无水乙醇100 mL,加沸石2~4颗,密塞,静置1 h,连接回流装置,90 ℃水浴回流1 h(沸腾后开始计时),关闭水浴锅电源并加入冷水降温后,迅速取下平底烧瓶,及时密塞,流水冷却至室温后,在真空度−0.01~−0.03 MPa下抽滤,抽干后每次用25 mL无水乙醇洗涤平底烧瓶、滤器和滤渣,共3次,滤液转移至已干燥至恒定质量的蒸发皿中,在90 ℃水浴上蒸干后,于105 ℃干燥3 h,置干燥器中冷却30 min,迅速精密称定质量,以干燥物来计算醇溶性浸出物含量。结果显示15批样品的醇溶性浸出物质量分数为2.45%~6.39%,其中S13(山东聊城)含量最高。
2.2 比色法测定多糖含量
取“2.1”项下的水溶性浸出物,参照《中国药典》2020年版中灵芝多糖的含量测定方法(硫酸蒽酮法测定)[1],以葡萄糖质量分数为横坐标(),峰面积为纵坐标(),制定标准曲线为=7.095 4+0.037 8,2=0.999 9,计算供试品溶液中葡萄糖的含量,结果见表2。15批样品均符合《中国药典》2020年版要求,质量分数范围为1.04%~3.19%,其中样品S11(陕西汉中泸农1号)的多糖含量最高。
2.3 柱前衍生高效液相色谱法(PMP-HPLC)测定多糖含量[20]
2.3.1 对照品衍生化溶液的制备 取甘露糖、葡萄糖醛酸、半乳糖、岩藻糖各约10 mg,葡萄糖约100 mg,精密称定,用纯净水溶解并定容至50 mL量瓶中,摇匀,作为混合对照品溶液备用;取对照品来苏糖约10 mg,精密称定,用纯净水溶解并定容到25 mL,摇匀,作为内标对照品溶液备用;精密移取0.125 mL混合对照品溶液和0.125 mL内标,混匀。加入0.15 mol/L的NaOH溶液0.3 mL和0.1 mol/L的PMP溶液0.5 mL,充分混匀后,70 ℃水浴30 min,冰浴终止反应,加入0.15 mol/L的盐酸0.32 mL和纯净水0.65 mL,充分混匀后,12 000 r/min室温离心10 min,吸取上清液适量即得。
2.3.2 供试品衍生化溶液的制备 将“2.1”项下8 g灵芝子实体的水提物,定容至10 mL,经0.22 μm滤膜过滤后,取0.25 mL,加入0.250 mL三氟乙酸溶液,充氮气,酒精喷灯封管,置110 ℃烘箱中水解4 h,取出,放至室温;加入0.5mL甲醇,60 ℃以下真空减压干燥,反复处理3次,至水解液干燥完全;残渣精密加入0.125 mL热水和0.125 mL内标(来苏糖),超声30 s,使残渣完全溶解并与内标充分混匀,加入NaOH 0.3 mL和PMP 0.5 mL,充分混匀后,70 ℃水浴保温30min,迅速取出,冰浴30 s终止反应,加入0.32 mL的HCL和0.65 mL 纯净水,充分混匀后,12 000 r/min室温离心10 min,小心吸取上清液适量,即得。
2.3.3 色谱条件 色谱柱Waters Symmetry Shield RP18(250 mm×4.6 mm,5 μm),柱温30 ℃,以18%乙腈-82% 0.1 mol/L醋酸铵水溶液为流动相等度洗脱,体积流量为1 mL/min,检测波长250 nm,进样量20 μL,混合对照品色谱图和供试品衍生化溶液色谱图见图1。
2.3.4 方法学考察 精密度、稳定性、重复率、加样回收率试验根据文献方法[20]操作,RSD均小于3%,方法学考察结果符合要求。
2.3.5 样品的测定将所测供试品单糖按以下公式计算含量(计算时需扣除子实体的水分),单糖含量见表3。合计后得到多糖含量,结果见表2。多糖含量在0.33%~0.95%,均符合《中国药典》2020年版规定,其中S13(山东聊城)含量最高。
单糖量=(u/s)×s×(/)×100
u为样品溶液中目标物与内标物的峰值响应比,s为标准溶液中目标物与内标物的峰值响应比,s为经衍生化的标准溶液等分试样中相关分析物的量,为相对于样品溶液体积的稀释系数,为制备样品溶液所用灵芝子实体的质量
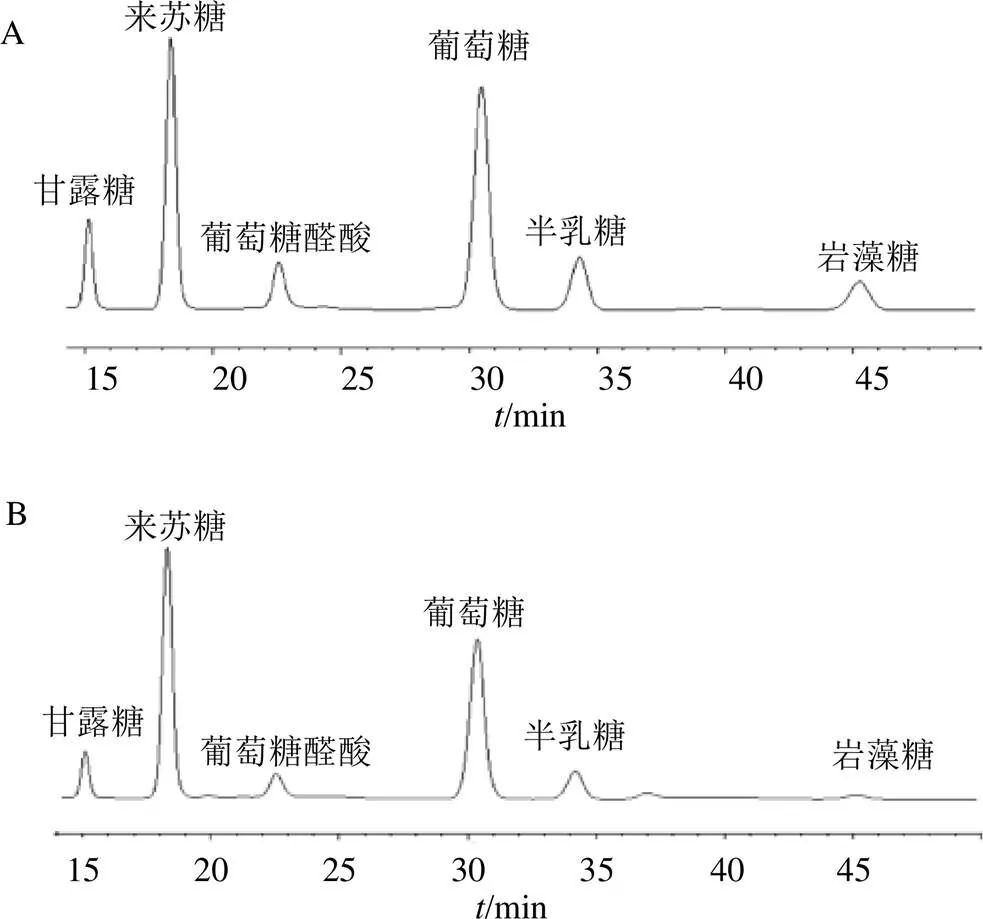
图1 混合对照品(A)和供试品(B) 衍生化溶液色谱图
2.4 高效凝胶渗透色谱法(HPGPC)测定多糖含量[21]
不同来源或种类的灵芝,其多糖成分的相对分子质量有较大差异,HPGPC法作为测定多糖相对分子质量及含量的常见分析手段,已经成为灵芝质量评价的行业共性方法,本实验也将其纳入不同产地赤芝质量考察的标准之一。
2.4.1 混合对照品溶液的制备 取右旋糖酐及葡萄糖对照品约50 mg,精密称定,分别加超纯水溶解,定容至5 mL量瓶中,制成10 mg/mL的右旋糖酐对照品储备液和葡萄糖对照品储备液。混合对照品参数见表4。
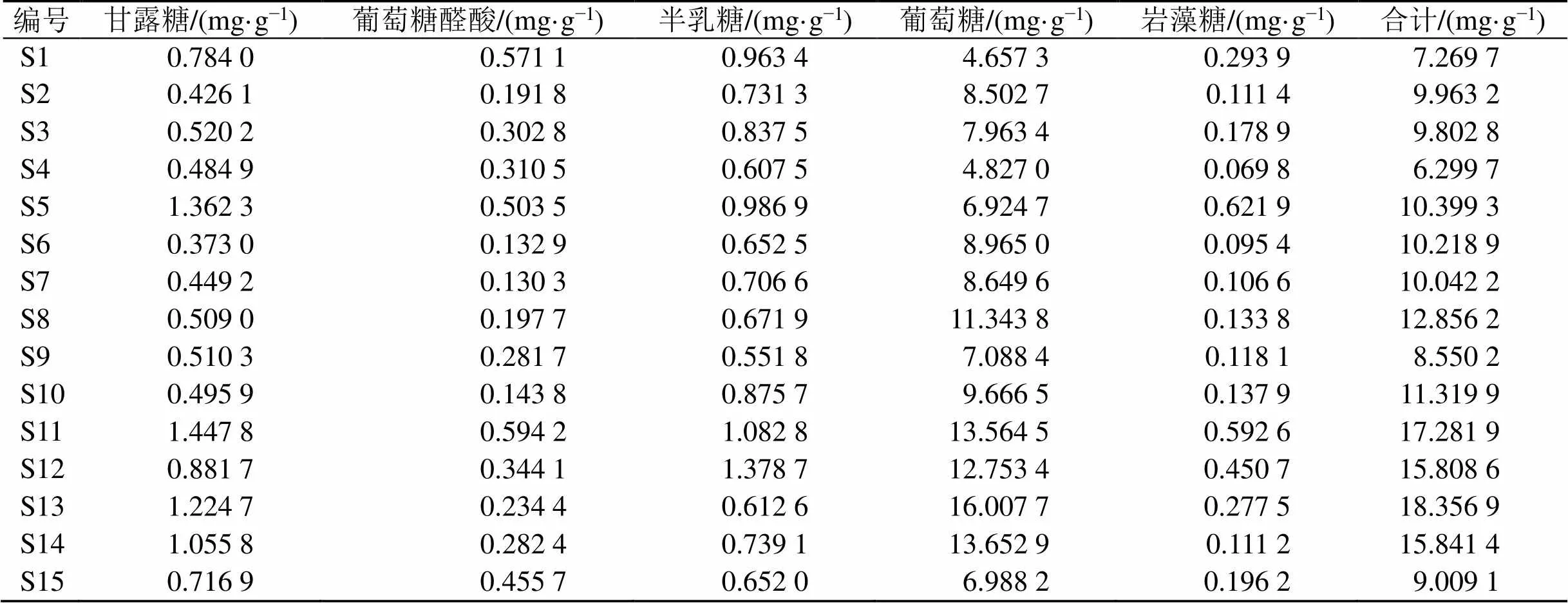
表3 15批赤芝中单糖含量

表4 右旋糖酐对照品参数
2.4.2 供试品溶液的制备 取“2.1”项下的水溶性浸出物滤液160 mL(约2 g样品),沸水浴蒸干,用5 mL热水超声溶解,缓慢加入无水乙醇至总体积约40 mL,超声,4 ℃下静置12 h,4000 r/min,4 ℃离心30 min,弃上清液后真空干燥,沉淀物用热水溶解并转移至10 mL量瓶中,密塞封口,50 ℃超声10 min,放至室温,定容至刻度线,摇匀,取溶液约5 mL,室温下4000 r/min离心10 min,上清液过0.45 μm微孔滤膜,取3 mL左右续滤液备用。
2.4.3 色谱条件 采用串联凝胶色谱柱TSK-GEL(300 mm×7.8 mm,10 μm),柱温35 ℃;流动相为0.71% Na2SO4硫酸钠水溶液,体积流量0.5 mL/min,进样量20 μL。
2.4.4 标准曲线绘制 以各对照品峰位相对分子质量的对数作为纵坐标(),峰顶时间作为横坐标(),凝胶渗透色谱专用软件校正标准曲线得=10.491 4−0.162 4,2=0.998 2。
2.4.5 方法学考察 精密度、稳定性、重复性、加样回收率试验按照参考文献方法[21]操作,RSD均小于3%,方法学考察结果符合要求。
2.4.6 样品测定 将值设为5000和400 000代入标准曲线公式,求得对应峰位相对分子质量下的峰顶时间1和2,对各供试品图谱1到2之间的响应面积进行积分,获得峰位相对分子质量5000~400 000的响应面积S1;积分标识相对分子质量为5000~670 000的右旋糖酐对照品图谱,获得各进样对照品的对应峰面积,并统一折算成10 mg/mL浓度的峰面积,然后求得平均峰面积S2。代入下式求得多糖含量,具体测定结果见表2。结果表明15批赤芝药材多糖含量在0.72%~2.51%,其中赤芝S11(陕西汉中泸农1号)的多糖含量最高。
多糖含量=S1/S2×10×10/子实体质量/1000
2.5 比色法测定总三萜及甾醇含量[1]
2.5.1 对照品溶液的制备 取干燥至恒定质量的齐墩果酸对照品约10 mg,精密称定,加甲醇溶解并定容至50 mL量瓶中,制成含齐墩果酸0.2 mg/mL的对照品储备液。
2.5.2 供试品溶液的制备 灵芝子实体打粉,过筛,取2~5号筛之间的粉末约2 g,精密称定,置150 mL具塞锥形瓶中,精密加入无水乙醇100 mL,加沸石2~4颗,密塞,称定质量,静置1 h,水浴回流1 h,冷却至室温,称定质量,用无水乙醇补足失重,摇匀,取适量过0.22 μm微孔滤膜,取续滤液约2 mL备用。
2.5.3 标准曲线的绘制 精密量取对照品溶液0.1、0.2、0.3、0.4、0.5 mL,分别置15 mL具塞试管中,挥干,预冷2 min后精密加入新配制的香草醛冰醋酸溶液(精密称取香草醛0.5 g,加冰醋酸使溶解成10 mL,即得)0.2 mL,高氯酸0.8 mL,具塞,从冰浴取出,涡旋混匀,移入70 ℃水浴中加热15 min,立即取出置冰浴中冷却5 min,取出置室温水中回温5 min,精密加入醋酸乙酯4 mL,涡旋混匀,采用石英比色皿,以相应试剂为空白,照紫外-可见分光光度法(通则0401)[19],在546 nm波长处测定吸光度,以吸光度为纵坐标(),浓度为横坐标(),绘制标准曲线=8.095 4+0.038 8,2=0.999 9。
2.5.4 样品的测定 精密量取供试品溶液0.2 mL,置15 mL具塞试管中,照标准曲线制备项下的方法,自“挥干”起,同法操作,测定吸光度,从标准曲线计算供试品溶液中齐墩果酸的含量,结果见表2。15批赤芝都满足《中国药典》2020年版要求,范围在0.70%~1.74%,其中赤芝S13(山东聊城产)三萜甾醇含量最高。
2.6 HPLC法测定灵芝酸含量[22]
2.6.1 色谱条件 色谱柱Waters Symmetry Shield RP18(250 mm×4.6 mm,5 μm),体积流量1 mL/min,检测波长257 nm,进样量20 μL,柱温20 ℃。以0.1%磷酸水(A)-乙腈(B)作为流动相,洗脱程序为0~10 min,85%~75% A;10~58 min,75%~71% A;58~60 min,71%~69% A;60~68 min,69% A;68~78 min,69%~64% A;78~98 min,64% A;98~100 min,64%~0% A。
2.6.2 混合对照品溶液的制备 取灵芝酸A约4 mg,灵芝酸B、灵芝烯酸B、灵芝酸C2、灵芝烯酸C、灵芝烯酸D、灵芝酸D、灵芝酸F、灵芝酸G、灵芝酸H各约2 mg,精密称定,用甲醇溶解并定容到10 mL量瓶中,摇匀备用。制得各对照品溶液均为0.2 mg/mL的对照品溶液。
2.6.3 灵芝酸A对照品溶液的制备 取灵芝酸A对照品约10 mg,精密称定,用甲醇溶解并定容到25 mL量瓶中,配制成0.442 mg/mL的灵芝酸A的对照品原液,再逐步稀释为0.331 500、0.221 000、0.110 500、0.055 250、0.027 625 mg/mL的对照品溶液。
2.6.4 供试品溶液的制备 精密称定赤芝粉末3 g,加入150 mL醋酸乙酯,水浴回流1 h,抽滤,用25 mL醋酸乙酯洗涤3次,合并滤液,85 ℃水浴挥干,用甲醇5 mL溶解残渣并定容至5 mL量瓶中,摇匀,过0.22 μm微孔滤膜,备用。
2.6.5 标准曲线的绘制 将“2.6.3”项不同质量浓度的灵芝酸A对照品溶液分别进样,以质量浓度为横坐标(),以峰面积为纵坐标()绘制标准曲线,得=11 290.231 6+11.852 7,2=0.999 9。
2.6.6 方法学考察 精密度、稳定性、重复率、加样回收率试验根据参考文献方法[22],RSD均小于3%,方法学考察结果符合要求。
2.6.7 样品测定 将混合对照品和供试品溶液分别进样,色谱图见图2。以灵芝酸A标准曲线进行计算,其他目标峰按换算因子全部折算为灵芝酸A,求和即得灵芝酸总量,结果见表2。结果表明15批赤芝药材灵芝酸含量在0.05%~0.79%,其中山东聊城产赤芝S4、S9的灵芝酸含量较高。
灵芝酸量=(u/s)×s×(/)×100
u为样品溶液中目标物的峰面积,s为标准溶液中灵芝酸A的峰面积,s为标准溶液中灵芝酸A的浓度,为制备样品溶液的灵芝子实体质量,为与灵芝酸A的相对响应因子
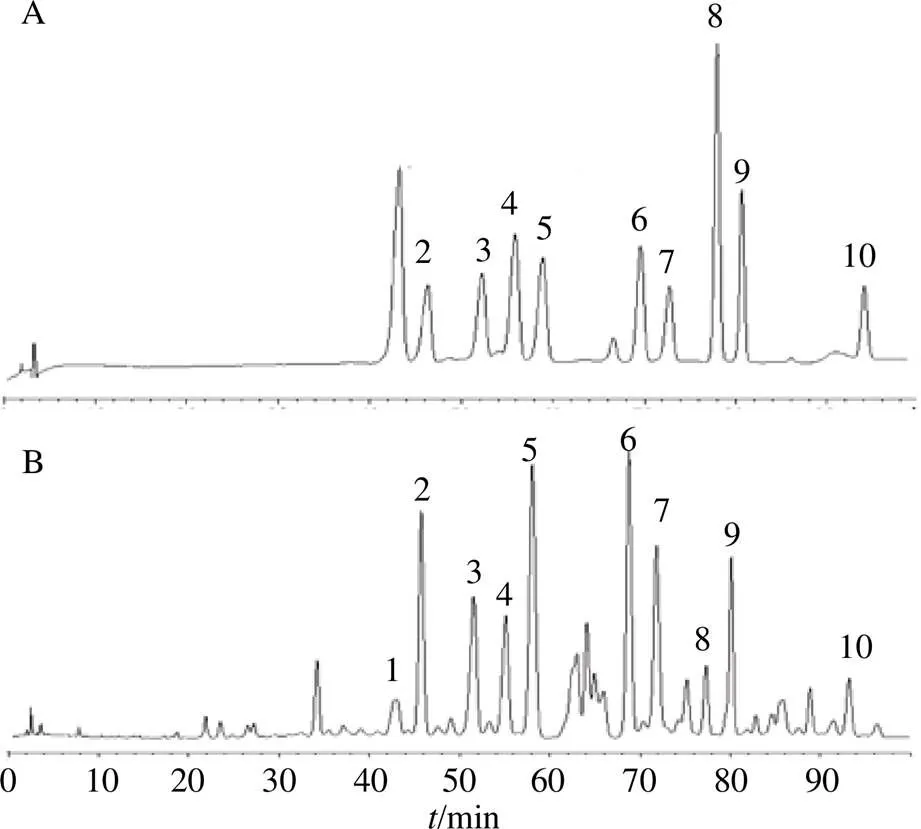
1-灵芝烯酸C 2-灵芝酸C2 3-灵芝酸G 4-灵芝烯酸B 5-灵芝酸B 6-灵芝酸A 7-灵芝酸H 8-灵芝烯酸D 9-灵芝酸D 10-灵芝酸F
2.7 指纹图谱的建立
2.7.1 供试品溶液的制备 取灵芝细粉约3 g,精密称定,加入250 mL洁净干燥平底烧瓶,精密加入150 mL醋酸乙酯,加沸石2~3颗,具塞,静置浸泡1 h,水浴回流1 h,抽滤,用25 mL醋酸乙酯洗涤平底烧瓶、滤纸和滤渣,重复3次,滤液入洁净干燥蒸发皿,水浴挥干,用5 mL甲醇溶解残渣并完全转移至5 mL量瓶中,定容,摇匀,过0.22 μm微孔滤膜,备用。
2.7.2 混合对照品溶液的制备 同“2.6.2”项
2.7.3 色谱条件 同“2.6.1”项。
2.7.4 方法学考察 同时取赤芝S15的供试品溶液进行精密度、重复性和稳定性的验证,各项RSD均小于3%,方法学考察结果符合要求。
2.7.5 指纹图谱的建立 将15批赤芝的指纹图谱导入《中药色谱指纹图谱相似度评价系统》(2012版),以赤芝S15指纹图谱作为参考谱图,进行峰匹配,以中位数生成对照图谱,得到15批赤芝指纹图谱和对照指纹图谱见图3,共得到23个共有峰,同时对指纹图谱进行相似度评价,结果见表5。

2-灵芝烯酸C 3-灵芝酸C2 5-灵芝酸G 6-灵芝烯酸B 8-灵芝酸B 12-灵芝酸A 15-灵芝酸H 17-灵芝烯酸D 19-灵芝酸D 23-灵芝酸F

表5 15批赤芝指纹图谱相似度
以共有峰的峰面积为变量,将15批赤芝的峰面积经归一化处理后,进行主成分分析(principal component analysis,PCA),自动拟合得到4个主成分,2为0.969,2为0.57(>0.5),模型拟合较好且有良好的预测性,结果见图4。15批赤芝分成了3组,山东聊城的赤芝(S4、S9、S13)与其余批次的赤芝差异较大。
根据PCA的结果,将15批赤芝分成2组,一组为山东聊城组,一组为其他产地,进行正交偏最小二乘判别分析(orthogonal partial least squares discriminant analysis,OPLS-DA)(图5),自动拟合得到6个主成分,2为0.936,2为0.96。选择VIP≥1的色谱峰作为区分山东聊城与其他产地赤芝的标志差异物(色谱峰以保留时间计),VIP值见图6,其中VIP≥1的色谱峰有8个,分别为峰19(灵芝酸D)、18、4、9、15(灵芝酸H)、23(灵芝酸F)、21、5(灵芝酸G)。

图4 15批赤芝PCA图
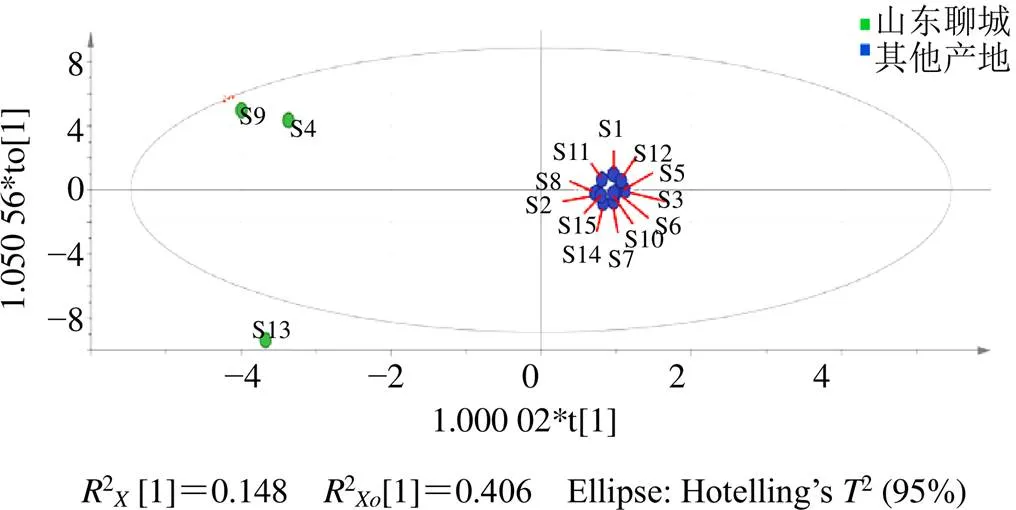
图5 15批赤芝OPLS-DA图
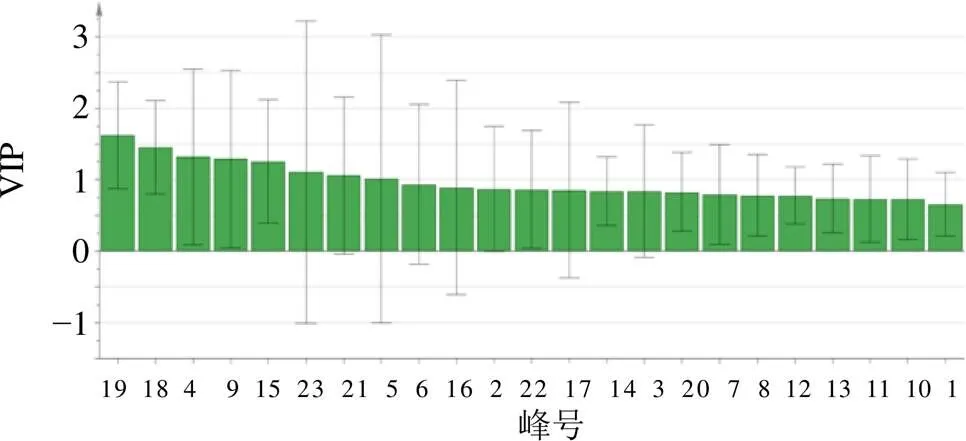
图6 15批赤芝的VIP图
3 讨论
本实验对15批不同产地和批次的赤芝,进行了全面质量控制方法考察,15批赤芝全部符合《中国药典》2020年版的要求。灵芝产业庞大,市面上存在跟风种植的情况,针对未来中药材产业升级以及绿色农业的趋势,国内需要加快GAP的建设,中国作为灵芝药材大国和市场需求大国,灵芝GAP基地更应该以严格的质量标准,逐步规范化、标准化,不断推动灵芝产业的发展。
对比3种多糖质量评价方法的结果可知,测定方法不同,测定结果有显著差异。灵芝多糖种类繁多,结构复杂,是由3股单糖链构成的具有螺旋状立体构型(三维结构)的葡聚糖[23],主要由葡萄糖、半乳糖、甘露糖、岩藻糖、木糖和阿拉伯糖通过不同比例和不同糖苷键类型连接构成,活性多糖有效成分附着在细胞壁内侧,大部分直接与蛋白质形成分子络合物[24],而3种多糖检测方法都是基于醇沉除杂工艺,因此非糖类物质容易与目标多糖同步沉淀成为代测物。硫酸蒽酮法测定总多糖,原理是氧化出醛基与蒽酮结合并显色,在固定波长下检测,这些非糖类化合物会参与显色反应,或本身在测定波长下以强吸收的方式形成干扰误差。HPGPC法的原理是基于不同分子量在凝胶色谱柱中的分离情况,因此待测定物的计算也会受到杂质干扰。总之,可以综合3种多糖的考察方式,视实际情况选择评价手段。
对灵芝药材中三萜类成分进行含量测定结果表明,甾醇作为灵芝最重要的活性成分之一,是灵芝质量的重要指标,《中国药典》测定灵芝三萜和甾醇,但由于灵芝三萜提取率低、杂质多,紫外分光光度法的测定容易受到提取物中皂苷、油酸等物质干扰,导致特异性不强,三萜甾醇含量与实际有较大的差异;目前从灵芝中已分离出300多种三萜类成分[25],其中灵芝酸是最主要的成分,针对三萜中的灵芝酸,以灵芝酸A来计算10种灵芝酸含量,该测定物质较明确,但未将甾醇部位纳入灵芝活性成分的考察。
指纹图谱的PCA分析和OPLS-DA分析结果中山东聊城的S13、S4、S9与其他批次的差异较大,其中S13中三萜甾醇含量最高,灵芝酸含量最低,可能是S13中中性三萜和甾醇等脂溶性成分的含量更高;S4、S9中灵芝酸含量较高,三萜甾醇含量居中。
山东聊城GAP基地赤芝在三萜或甾醇等脂溶性成分上区别于其他产地批次的赤芝,经VIP分析得到8个与其他批次不同的主要色谱峰,可见灵芝酸类成分在区分山东聊城和其他产地的灵芝上具有重要意义,同时挖掘指纹图谱的标志差异峰也可以为不同产地灵芝的质量评价提供依据。
利益冲突 所有作者均声明不存在利益冲突
[1]中国药典 [S]. 一部. 2020: 195-196.
[2]Wu H H, Tang S S, Huang Z Q,. Hepatoprotective effects and mechanisms of action of triterpenoids from Lingzhi or reishi medicinal mushroom(Agaricomycetes) on α-amanitin-induced liver injury in mice [J]., 2016, 18(9): 841-850.
[3]Ahmad M F.: A rational pharmacological approach to surmount cancer [J]., 2020, 260: 113047.
[4]Cao Y, Xu X W, Liu S J,.: A cancer immunotherapy review [J]., 2018, 9: 1217.
[5]Kuok Q Y, Yeh C Y, Su B C,. The triterpenoids ofprevent stress-induced myocardial injury in mice [J]., 2013, 57(10): 1892-1896.
[6]Yu N H, Huang Y P, Jiang Y,.triterpenoids (GLTs) reduce neuronal apoptosis via inhibition of ROCK signal pathway in APP/PS1transgenic Alzheimer's disease mice [J]., 2020, 2020: 9894037.
[7]Wang C F, Liu X M, Lian C L,. Triterpenes and aromatic meroterpenoids with antioxidant activity and neuroprotective effects from[J]., 2019, 24(23): 4353.
[8]Jiang G Y, Lei A T, Chen Y,. The protective effects of thepolysaccharide against acrylamide-induced inflammation and oxidative damage in rats [J]., 2021, 12(1): 397-407.
[9]Li L, Xu J X, Cao Y J,. Preparation ofpolysaccharide‑chromium (III) complex and its hypoglycemic and hypolipidemic activities in high-fat and high-fructose diet-induced pre-diabetic mice [J]., 2019, 140: 782-793.
[10]Xiao C, Wu Q P, Xie Y Z,. Hypoglycemic mechanisms ofpolysaccharides F31 in db/db mice via RNA-seq and iTRAQ [J]., 2018, 9(12): 6495-6507.
[11]Ko H H, Hung C F, Wang J P,. Antiinflammatory triterpenoids and steroids fromand G. tsugae [J]., 2008, 69(1): 234-239.
[12]Ren L, Zhang J, Zhang T H. Immunomodulatory activities of polysaccharides fromon immune effector cells [J]., 2021, 340: 127933.
[13]Wang J, Cao B, Zhao H P,. Emerging roles ofin anti-aging [J]., 2017, 8(6): 691-707.
[14]Bishop K S, Kao C H J, Xu Y Y,. From 2000years ofto recent developments in nutraceuticals [J]., 2015, 114: 56-65.
[15]Gong T, Yan R Y, Kang J,. Chemical components of[J]., 2019, 1181: 59-106.
[16]Baby S, Johnson A J, Govindan B. Secondary metabolites from[J]., 2015, 114: 66-101.
[17]邢佳慧. 灵芝属的物种多样性、分类与系统发育研究 [D]. 北京: 北京林业大学, 2019.
[18]姜涛, 施枝江, 姚艺新, 等. 不同品种灵芝多指标的质量评价研究 [J]. 中药材, 2018, 41(12): 2847-2855.
[19]中国药典 [S]. 四部. 2020:114-115, 232.
[20]王浩豪, 戴军, 陈尚卫, 等. 灵芝孢子粉多糖的PMP-HPLC指纹分析[J]. 食品与发酵工业, 2011, 37(12): 133-136.
[21]赛贞, 林树钱, 林志彬, 等. 高效凝胶渗透色谱法测定双灵固本散中多糖肽的峰位分子量 [J]. 中成药, 2007 (9): 1386-1388.
[22]罗舒, 宋怡, 罗霞, 等. 灵芝中10种灵芝酸类成分含量一测多评法的建立 [J]. 中国药房, 2023, 34(14): 1703-1706.
[23]Synytsya A, Novák M. Structural diversity of fungal glucans [J]., 2013, 92(1): 792-809.
[24]Kumakura K, Hori C, Matsuoka H,. Protein components of water extracts from fruiting bodies of the reishi mushroomcontribute to the production of functional molecules [J]., 2019, 99(2): 529-535.
[25]Xia Q, Zhang H, Sun X. A comprehensive review of the structure elucidation and biological activity of triterpenoids from. [J]., 2014, 19(11): 17478-17535.
Quality evaluation offrom different areas
ZENG Miao, XIE Meng-jun, ZHU Zi-yu, YU Yue-ting, ZHANG Mei
State Key Laboratory of Southwestern Chinese Medicine Resources, School of Pharmacy, Chengdu University of Traditional Chinese Medicine, Chengdu 611137, China
A comprehensive quality evaluation analysis of Chizhi [(Leyss. exFr.) Karst.]samples from different batches or origins was conducted to provide a basis for the improvement of the quality standard of Lingzhi () and to screening of high-qualitymaterials.Referring to the method of2020 edition and common approaches in this field, fifteen batches ofherbs from each GAP base were collected and examined for moisture, ash, water-soluble leachate, alcohol soluble leachate, the polysaccharide content was compared by determining colorimetric method, precolumn derivatization high performance liquid chromatography (PMP-HPLC), and high performance gel permeation chromatography (HPGPC). The triterpene sterol content was determined by colorimetric method, and the QAMS of ganoderic acid A was evaluated by HPLC method.. The fingerprint profiles of 15 batches ofwere also compared for differences.All 15 batches ofmet the quality standard requirements of the2020 edition, the results of fingerprint cluster analysis and principal component analysis (PCA) show thatof Liaocheng, Shandong Province can be clearly distinguished from other batches of.All 15 batches ofmet the quality standards of the, indicating that the quality of the domesticGAP base cultivation is good; at the same time,of Liaocheng, Shandong Province can be significantly different from other origin batches ofsamples, mainly in the higher content of active ingredients such as triterpene sterols.
(Leyss.exFr.) Karst.; quality evaluation; principal component analysis; polysaccharides; triterpenes; fingerprint
R286.2
A
0253 - 2670(2023)21 - 7193 - 09
10.7501/j.issn.0253-2670.2023.21.028
2023-02-10
国家自然科学基金资助项目(81774202);成都中医药大学杏林学者学科人才科研提升计划(CXTD2018010)
曾 秒,女,硕士,研究方向为药物分析。E-mail: 1220799848@qq.com
通信作者:张 梅,女,博士生导师,从事中药药效物质基础及质量控制与评价研究。E-mail: zhangmei63@cdutcm.edu.cn
[责任编辑 时圣明]
