含能化合物量子化学数据库的设计及应用
2023-11-07张朝阳
黄 鑫,张朝阳
(中国工程物理研究院化工材料研究所,四川 绵阳 621999)
0 引 言
为满足现代武器对含能化合物综合性能的高需求,科研人员以实验探索与理论计算的方式研究高性能含能化合物,产生了大量极具价值的包括含能化合物设计、合成、表征在内的数据[1-4]。例如,含能化合物的分子模拟研究能够获得包括几何结构、电荷分布、热力学性质、爆炸/分解反应路径以及基于定量构效关系(QSPR)的性能预测模型等[5-6];含能化合物的合成与表征研究能够获得包括化学反应路径与机理、分子/晶体结构、能量安全特性、力热性质等数据[7-9];含能化合物在武器装药中涉及到配方设计和评估等研究,涵盖了包括黏合剂、增塑剂、键合剂、安定剂、钝感剂和工艺助剂等物质的相关性能数据[10]。
这些含能化合物及其相关物的实验与计算数据分散在各种报告、期刊、专利、书籍、特殊文献中,数据收集与数据质量甄别困难较大;且出于数据敏感性与涉密性的原因,现有的含能化合物实验数据库通常只向特定的组织和人员开放访问权限,如北约弹药安全信息分析中心的Energetic Materials Compendium(EMC)数据库[11]以及德国ICT 热化学数据库等。尽管在医药[12]、化学化工[13]、能源与金属材料[14]等开放研究的热点领域已经建立了规模较大的量子化学计算数据库并实现了较高程度的数据开放共享,目前国内外尚缺少专门的数据库用于收集含能化合物量子化学计算产生的数据,以供研究人员获取与使用。随着数据驱动下的材料智能设计时代的到来,含能化合物的量子化学理论计算、高通量虚拟筛选技术等愈发成熟,能够实现对含能化合物的结构与性质进行高精度的分析和预测。在此基础上建立含能化合物量子化学数据库,收集含能化合物在高精度计算水平下的量子化学数据,既能够避免大量重复性的计算研究与资源消耗,也保证了数据质量以便于进行深入分析及知识挖掘。
量子化学计算研究含能化合物能够获得的数据涵盖面广并且针对特定的能量安全性质,很难进行详尽的列举,感兴趣的研究人员可以参考Peter Politzer等[15]以及肖鹤鸣教授课题组[16-17]出版的含能化合物理论计算与设计专著。量子化学计算是一种包含必要物理过程的严格方法,能够提供含能化合物分子设计的微观尺度信息,因此选择高精度的含能化合物量子化学计算数据、建立含能化合物量子化学计算数据库,对于含能化合物的智能分子设计具有重要意义。
本文主要总结并梳理近年来量子化学计算所获得的含能化合物关键性结构和性质数据种类、数据库与高通量虚拟筛选相结合的含能化合物分子设计,以期为含能化合物量子化学计算数据的产生与标准化制定、数据库的概念设计及潜在的实际应用提供有益的参考。此外,以本课题组开发的含能化合物量子化学高通量计算平台(EM Studio)与含能化合物量子化学数据的数据库(EM Database)为例,提供含能化合物量子化学数据从产生、收集与开放共享的具体案例。
1 含能化合物的量子化学计算数据
数据质量是数据库建设的重点工作,包括数据的准确性、合规性、完整性、及时性、一致性等维度。量子化学计算基于量子力学的基本原理和方法研究化学问题,通过对物理过程的精确计算和预测得到材料的性质。含能化合物结构和性能的研究不仅涉及到从常规状态到高温高压的极端条件,也涉及到从基态到快速反应的燃烧和爆轰过程。量子化学计算作为理解、预测以及设计含能化合物的基础方法,其准确性对于所生成数据的有效性极为重要。从头算和半经验方法、密度泛函方法等均在含能化合物的研究中获得应用,其中基态下分子与晶体的结构与性质研究最为基础。研究人员也提出了基于量子化学计算结果的定量构效关系模型,例如基于表面静电势的密度校正模型[18]、基于等键反应的生成焓计算模型[19]、基于引发键解离能的感度预测模型[20]等。适用于含能化合物(包括共价、离子化合物等)的量子化学计算理论方法与性质预测模型不同,其中对含有CHNO 元素的中性分子的方法发展较为成熟。下面以含有CHNO 元素的中性含能分子为例,梳理量子化学计算所能够得到的基础量子化学计算数据。
1.1 计算方法
目前,研究人员广泛使用包括GAUSSIAN、ORCA、VMD、Multiwfn 等程序软件完成含能分子的量子化学计算与结果处理。借助统计热力学理论,可以获得含能分子在不同温度下的性质参数,如焓、熵、自由能、生成热、比热等性质。由于密度泛函方法的结果可靠、计算耗时较低,因此在含能化合物的结构优化、振动分析以及热力学性质计算方面获得了广泛使用。密度泛函方法的泛函与基组选择对于计算耗时以及结果的准确性有重要影响,研究人员对计算方法的选择并没有统一标准。例如,广泛使用的泛函包括交换相关泛函PBE、杂化泛函B3LYP、PBE0、M06 系列,经验弥散泛函wB97XD 等;对基组的选择则有Pople系列基组以及Dunning 相关一致性基组等。而对于某些热力学参数进行高精度的计算,则需要使用组合方法,例如CCSD(T)外推至CBS 完备基组方法以及Gaussian-4(G4)组合方法等。
1.2 几何结构数据
分子的几何结构数据主要记录了分子中每个原子的元素种类和三维空间区域中的坐标值,可以进一步得到键长、键角、二面角、分子密度、体积、表面积等信息。其记录格式有多种,能够被计算化学软件读取的通用文件格式包括xyz 文件格式、pdb 文件格式、mol文件格式等。
1.3 电子结构数据
密度泛函计算方法将电子密度作为最基本的参量,用于描述和确定分子体系的性质。通过电子密度能够与势能及能量有关的性质建立关联,包括前线轨道能级(最高占据轨道能量EHOMO、最低未占据轨道能量ELUMO、能级差)、化学键级、原子电荷、分子极矩、电离能等。
1.4 反应性数据
含能分子的反应性包括热稳定性、机械感度等。对于热稳定性而言,键解离能的数值代表了化学键的强弱,与热分解性质具有关联性。对于机械感度而言,分子的静电势反映了分子的电荷分布、极值点以及正负电荷分离的情况,而不均衡的静电势分布往往导致亚稳定性与机械感度高。
1.5 热力学性质数据
热力学性质主要分为两类,其数值与计算所规定的热力学系综条件(温度和压力)有关,分别为在绝对零度条件下计算得到的分子生成焓、零点振动能、焓值、吉布斯自由能,以及经过温度和压力校正后的特定温度与压力条件下的上述数据。
1.6 谱学性质数据
密度泛函计算方法能够得到包括基态和激发态的谱学性质。例如,使用微扰理论方法能够得到包括红外、Raman 在内的振动光谱数据以及包括NMR 在内的磁谱数据;而使用电子/中子激发计算则能够获得非平衡态的谱学性质数据。
由此可见,量子化学以及结果的进一步处理计算能够获得种类丰富的数据信息。这也对数据的产生与收集提出了具体的要求:首先,计算方法可靠性的验证是保证数据质量的前提,需要在计算研究中选择具有鲁棒性的方法以获得有意义的数据;其次,数据的收集需要设计专用的表结构与编码规则,实现标准化与规范化。
2 数据库与高通量虚拟筛选相结合的含能化合物智能设计
2.1 含能化合物数据库
数据库是为满足具体的信息要求而设计的一个逻辑相关数据及其描述的共享集。数据库含有大量数据集、能满足多用户同时使用。除大量的纸质印刷版数据集手册外,现阶段分子与材料的数据库主要为可开放获取的网络资源,表1 汇总了其中的部分数据库网络资源,其主要分为计算和实验两大类型。而从所收录的数据信息做区分,大致分为如下4 类:(1)计算模拟 数 据 库,包 括Materials Project、AFLOWlib、Pub-ChemQC、Open Quantum Materials Database(OQMD)等;(2)分子信息学库,包括GDB、ChEMBL、ChemSpider、PubChem 等;(3)晶体结构信息库,包括CSD、ICSD 等;(4)化学反应信息库,包括Reaxys、Sci-Finder、USPTO/Lowe 等[21]。与之相比,现有的含能化合物数据库数量有限且获取难度较高,目前能够公开获取的含能化合物性能数据主要集中在纸质印刷版数据手册中。表2 总结了部分含能化合物及其相关物综合性能的数据手册信息,其中收录的数据以分子或者晶体的实验性质结果为主,且不同手册的数据所采用测试标准不同、数值间差异化比较显著,数据质量的甄别困难较高。而基于分子模拟,尤其是量子化学计算的含能化合物数据集尚未见报道。
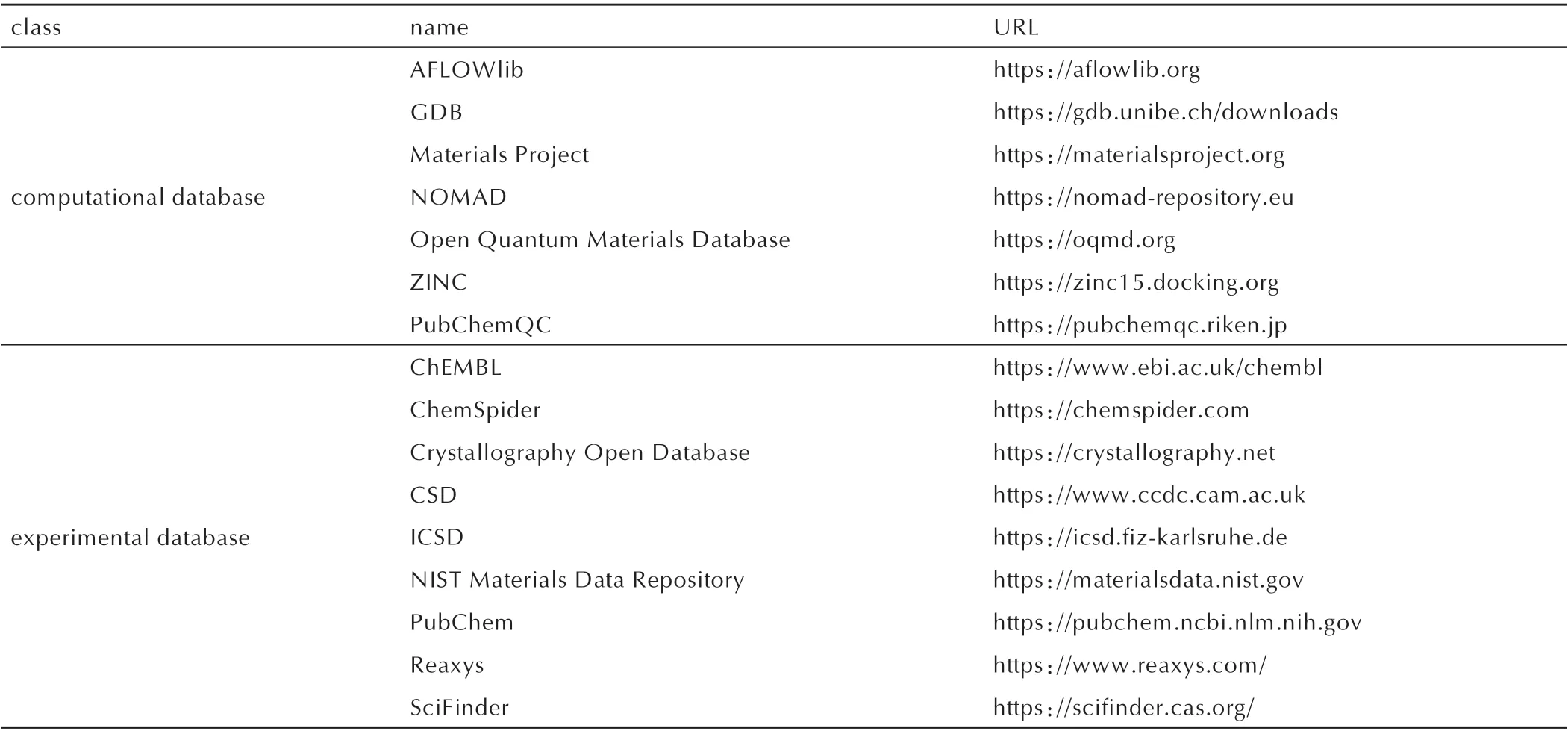
表1 可开放获取的分子与材料的数据库Table 1 Open access databases of molecules and materials

表2 部分含能化合物及其相关物综合性能的数据手册Table 2 Handbooks of properties of some energetic compounds and related materials
2.2 高通量虚拟筛选流程简介
材料传统的高通量筛选研究方式以实验为主,遵循与“设计-制造-测试-分析”的DMTA 循环模式类似的研发步骤,处理样品数量大,危险系数高、研究周期长、测试数据波动广且需要大量的资源投入;与之相比,高通量虚拟筛选能够以高效的方式对化合物的结构设计空间(~1026数量级)进行探索。结合特定的筛选标准与自动化技术,能够更进一步提升研发效率、缩短DMTA 循环周期,将化合物的设计效率推向新的高度。
高通量虚拟筛选流程通常由3 个步骤组成[30]:首先,基于电子结构以及热力学参数的计算获得包含材料性质的虚拟数据集;然后,通过合理的存储形式将这些性质信息在数据库系统中进行收集;最后基于所关注的特定性能对虚拟数据集进行统计分析或筛选、从中得到性质新颖的材料或者获得具有符合统计规律以及物理意义的新认识。必须指出的是,整个高通量虚拟筛选流程需要得到实验验证,以证明所构建的流程具有准确合理性。这样的反馈机制有利于构建更高质量的数据集以及提升筛选流程的预测能力与泛化性质。
2.3 数据库与高通量虚拟筛选相结合的含能化合物智能设计
含能化合物的高通量虚拟筛选研究,以含有CHNOF 元素的中性分子与晶体为主。由于有实验报道的含能化合物数量有限(不超过104),目前含能化合物虚拟数据集的构建方式主要有2 种:基于启发式的母体-取代基分子生成算法获得数据集、以及合并含能与非含能化合物的扩展数据集。基于启发式的母体-取代基分子生成工作方面,张朝阳课题组[31]从剑桥晶体数据库中收集并筛选了超过6 万种包含苯环结构的CHNO 分子的晶体结构,在结构拆分获得母体/取代基的基础上进行了分子生成,获得108数量级的潜在分子;在此基础上建立了基于生成焓、密度、键解离能以及分子平面度的筛选模型用于评估分子的性能(如图1a 所示)。结果表明,目标分子集合(A2)中六硝基苯是含苯环结构含能化合物中能量水平最高的而三氨基三硝基苯(TATB)具有最优的能量与安全综合性能。刘英哲等[32]以母体-取代基分子生成了约105数量级的含有CHNOF 元素的分子数据集,建立虚拟筛选模型最终获得综合性能的10 个潜在的含能化合物结构(如图1b 所示)。相似的母体-取代基研究思路也被用于设计零氧平衡的笼型骨架含能分子[33]。宋思维等[34]使用母体-取代基分子生成的模式获得约103数量级的含有氮杂环的分子数据集,建立虚拟筛选模型获得潜在的高能低感熔铸含能化合物。
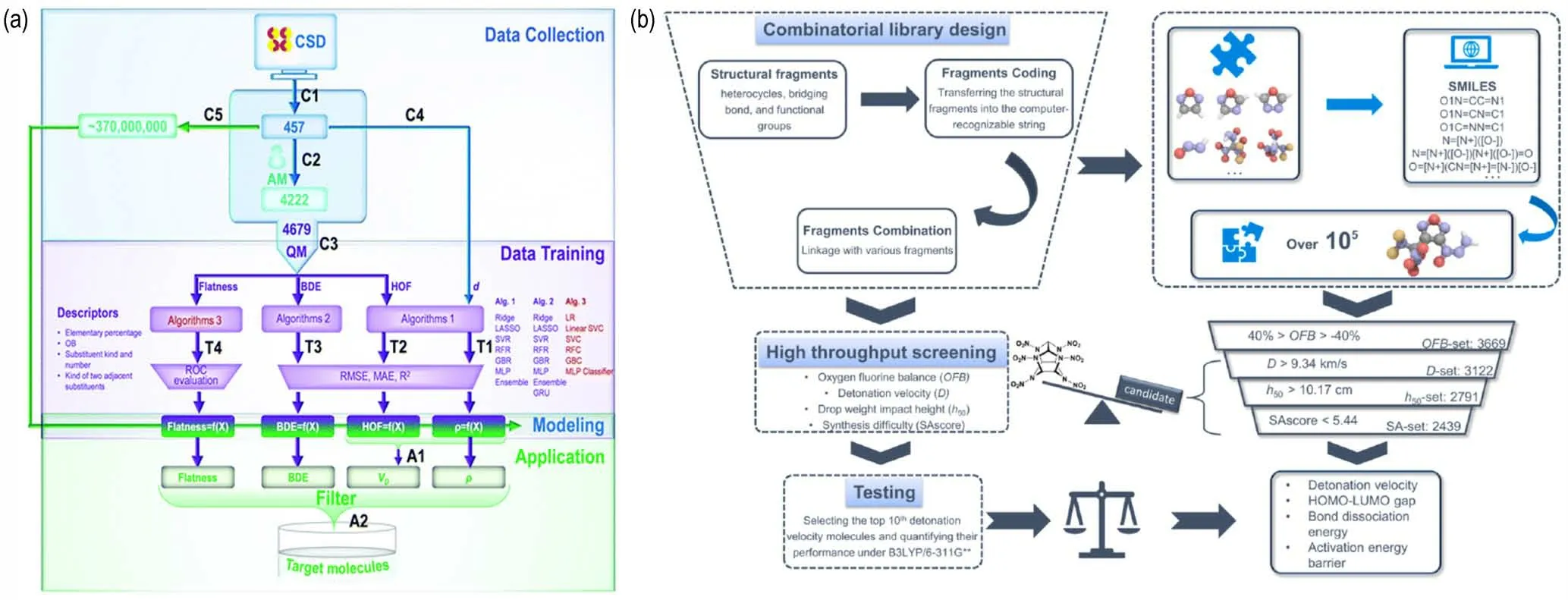
图1 基于启发式的母体-取代基分子生成(a)[31]以及含能材料高通量筛选(b)[32]Fig.1 Molecule generation works from heuristic base-substituent enumeration method(a)[31],and high-throughput screening of energetic materials (b)[32]
扩展数据集方面,麦吉尔大学的郭鸿课题组[35]收集了PubChem 数据库中的超过108个分子结构,使用高通量虚拟筛选获得了262 种超过1.5 倍TNT 当量的潜在含能化合物(图2a)。四川大学蒲雪梅课题组[36]从剑桥晶体数据库中获得了7871 种共晶的晶体结构数据(包括55 种含能共晶),使用图神经网络建立了虚拟筛选模型,并针对含能共晶进行了模型参数微调以达到更好的预测效果(图2b)。南洋理工大学的Li Shuzhou 课 题 组[37]也 开 发 了2 种 空 间 矩 阵 方 法,对PubChem 数据集中的CHNO 分子进行了晶体密度以及固相生成焓的筛选,并获得了56 种潜在的含能分子。

图2 基于扩展数据集的含能材料高通量筛选工作,包括PubChem 数据库(a)[35]以及剑桥晶体数据库(b)[36]Fig.2 High-throughput screening of energetic materials based on extended datasets,including PubChem database (a)[35]and Cambridge Crystallographic Data Centre (b)[36]
上述研究为含能化合物的设计提供了有益的研究思路,但也存在一定的局限性。首先,现阶段含能化合物的性能预估广泛使用经验模型获得预测参数,如密度、生成焓、爆速等;在虚拟筛选流程中可能存在经验模型的泛化性能不足,导致新型含能化合物性能预测结果有较大误差。其次,研究人员使用母体-取代基模式构建含能化合物数据集,所选用的取代基大多数为致爆基团,导致生成的化合物局限于种类有限的取代基,缺少结构的丰富性。此外,现阶段尚缺乏开放共享的含能分子结构数据集,也在一定程度上限制了含能化合物的分子生成、结构设计。
2.4 量子化学数据库结合高通量虚拟筛选的材料智能设计
量子化学计算能够获得电子结构、能量特性以及热力学性质的基础数据[38]。与实验数据相比,量子化学计算结果的可重复性好、易于批量化生成与数据开放共享。因此基于量子化学计算的高通量虚拟筛选已经在能源材料、医药等领域得到了应用。例如,Nicolas Mounet 等[39]从无机化学晶体结构数据库(ICSD)以及晶体开放数据库(COD)中收集了超过10 万个晶体结构,然后基于高通量的密度泛函计算筛选,从中获得了5619 种能够剥离出二维层状结构的母体材料。日 本 理 化 学 研 究 所RIKEN 的Maho Nakata 等[40-41]利用PM6 以及B3LYP/6-31G*方法计算了PubChem 数据库中收录的9100 万分子的几何结构以及HOMO-LUMO 能隙,并利用分子指纹谱以及机器学习算法得到了HOMO-LUMO 能隙的预测模型。上述领域的应用为含能化合物的量子化学虚拟筛选提供了有益的研究思路。
由此可见,构建含能材料专用的数据库是高通量筛选与智能设计的前提条件。从通用的化学与材料数据库中提取子结构、使用母体-取代基的分子生成构建虚拟的含能化合物的设计空间,是现阶段主要采用的2 种技术途径。然而上述方法存在较大的局限性:首先,含能化合物的能量与安全性能很少在通用数据库中收录,可开放获取的数据条目有限;其次,界定有机化合物为含能化合物的标准主要是含有致爆基的子结构或者基于分子结构的爆轰性质预测模型外推,这些筛选标准的可靠性需要进一步验证;此外,含能材料的量子化学计算是一种构建数据库的优势途径,数据包括分子以及晶体2 种体系,现阶段含能晶体结构的理论计算尚有待系统的方法验证并制定基准方法。
3 含能材料数据库的设计与应用展望
3.1 量子化学计算标准与数据模型
对于含能材料性能的实验测试,国内外均建立了较为系统的国家军用标准,对样品状态、测试方法以及数据收集的规范化提出了规定。对于含能化合物的量子化学计算,研究体系包括分子和晶体,研究人员使用的理论方法多样,且计算结果在文献中呈现方式以及必要数据条目的完备性差异大。量子化学计算数据库的建立,首先需要确定研究体系以及适用于该体系的理论方法与预测模型,进而选择精度高且成本低的计算方法,针对性地建立数据生成与格式化存储的标准与规范;其次,与化学与材料的通用数据库不同,含能材料数据库主要收录含能化合物的结构与性质数据,因此开发时需要对含能化合物进行界定,选择的标准包括分子的性质(包括元素组成、密度、爆速等),以及能量安全性质数值等。例如,对于元素类型仅限CHNO 的中性分子,使用B3LYP/6-31G(d,p)方法进行分子几何结构的优化,以及使用CBS-4M 方法进行分子能量的分析是目前比较可靠的量子化学计算方法;而使用K-J 方程也能够较为准确地获得爆轰性质数据[42]。
量子化学计算含能化合物能够获得种类丰富的性质数据(详见第1 部分),构建数据库需要对选择数据模型以结构化地组织与收录数据。关系数据模型以关系表的形式组织数据,具有很高的数据独立性,是目前数据库主流的数据模型。使用关系数据模型建立的数据库需要满足特定的规范,常见的关系数据库需要满足至第三范式的条件即可(即数据表不存在重复组(满足第一范式)、不存在部分依赖(满足第二范式)以及不存在传递依赖(满足第三范式))。
3.2 数据库设计
数据库设计包括概念结构设计与逻辑结构设计。关系数据库的设计通常使用实体(Entity)-联系(Relationship)的E-R 图对数据库进行概念结构设计。含能化合物与量子化学计算数据之间的联系包括一对一、一对多的情况。以CL-20 为例,由于─NO2的旋转存在多种稳定的分子构象,因此化合物名称和构象存在一对多的关系;而每一种构象与对应的几何结构数据等则存在一对一的关系。因此需要进行E-R 图对数据库进行逻辑结构设计,并在数据库中创建数据表、关系表及其他数据库对象。
含能化合物量子化学计算数据库的设计也要满足应用于数据的管理和检索,进而实现结构和性质的关系模型等数据应用的实际需求。数据的查询与获取功能包括分子结构的精确匹配查询、子结构查询、数值参数的查询、嵌套查询,查询结果的分组、排序、合并等。
3.3 数据库管理系统
为满足数据的管理和检索,常用的数据库管理系统以关系型数据库管理系统为主。数据库管理系统提供数据定义、数据操纵、数据完整性检查、数据安全保护、数据库存取与访问,并提供应用开发程序与数据库的接口。结构化查询语言(Structured Query Language,SQL)是用于关系数据库查询的结构化语言,其功能包括数据查询、数据操纵、数据定义和数据控制4 个部分。
关系数据库管理系统分为2 类:一类是桌面数据库,用于小型的单机应用程序,例如Access、FoxPro 和Excel 等;另一类是服务器数据库,主要适用于大型的多用户数据管理,包括Oracle、SQL Server、DB2、Sybase 等大型关系数据库管理系统,以及包括MySQL、PostgreSQL、SQLite 等小型关系数据库管理系统。这些常见的数据库管理系统能够实现格式化数据的增删改查操作与日常维护管理。
3.4 数据集扩展与应用技术开发
3.4.1 数据集扩展
含能化合物的能量与安全性质最受关注,因此这些性能数据有必要在作为量子化学计算结果的扩展,在数据库中收录。使用量子化学计算无法直接得到上述能量与安全性质的数据,包括爆速、爆压、机械感度等。因此需要采用后量化构效关系模型用于含能化合物多种性质的预测。构效关系模型所需的参数主要由量子化学计算产生的电子结构、波函数文件分析得到。
3.4.2 前端应用程序设计于开发
基于Java、PHP、VB/ASP.NET、Visual C#、Python等程序语言设计数据库前端应用程序,实现可视化界面设计、项目部署以及定制化的功能实现。
3.4.3 数据开放共享
量子化学计算产生的数据属于基础研究结果,且数据质量高、可重复性好,便于在公开平台实现较高程度的数据开放共享。笔者也希望以此种方式推动含能化合物研发的范式变革、加速研发效率、降低资源投入。
3.4.4 应用开发实例
针对含能化合物量子化学计算数据的产生、数据收集标准等问题,本课题组近年来分别开发并建立了能够在高性能计算集群上稳定运行的含能材料高通量计算交互式应用系统(Energetic Materials Studio,EM Studio)[43]用于实现含能化合物的高通量量子化学计算以及爆轰性质预测;此外,开发并建立了含能化合物量子化学计算数据采集与数据库管理系统(EM Database),用于结构化收集存储含有CHNO 元素的中性含能分子的量子化学计算数据以及爆轰参数(图3)。潜在含能分子结构主要是通过文献整理以及母体-取代基的方式完成,而对于分子含能与否的界定,则按照密度与爆速不亚于TNT 作为标准(即密度值1.648 g·cm-3,爆速值6950 m·s-1)。数据库中的数据也会随着计算方法的进步、更准确有效方法的确认而不断更新。
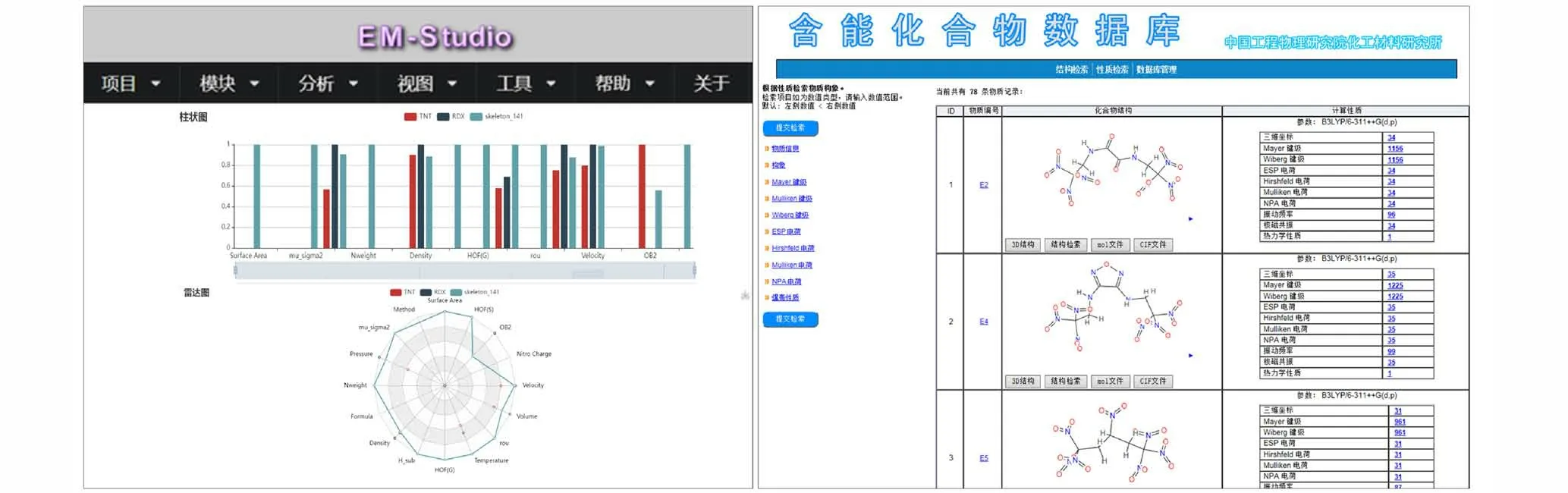
图3 EM Studio 以及EM Database 的应用程序界面[42]Fig.3 User interfaces of EM Studio program and EM Database program[42]
3.5 数据库的设计与应用展望
综上所述,数据库技术与含能材料量子化学计算的结合需要解决量子化学计算标准与数据模型、数据库设计、数据库管理系统选择、数据集扩展与应用技术开发等技术挑战。现阶段对于含有CHNO 元素的中性含能分子的量子化学计算以及爆轰性质预测方法可靠性高,易于通过高通量计算的方式进行分子设计与性质预测。此外,使用关系型数据库收录含能分子的结构与性质参数数据,以可视化用户界面的方式实现对数据库收录条目的开放获取也得到了实现。EM Studio 与EM Database 的实现证明了该技术方案的可行性。
未来含能化合物的数据库设计与应用需面向高性能含能化合物的实际需求,以高能、稳定、绿色为导向。在此对其设计与开发做以下两方面的展望:首先,含能化合物的性能与稳定性数据极为重要,但是现有预测模型的普适性仍需使用大量含能分子进行广泛验证,进而保证所产生数据的认可度。其次,数据库所收录的数据规模应尽可能大、性质条目尽可能全面,进而有利于含能化合物综合性能的设计。
4 结论与展望
本文总结了含能化合物的量子化学理论计算、高通量虚拟筛选技术、以及数据库技术。含能化合物的量子化学理论计算能够实现对含能化合物的结构与性质进行高精度的分析和预测。在此基础上建立含能化合物量子化学数据库,收集含能化合物在高精度计算水平下的量子化学数据,具有重要意义与实际价值。
含能化合物量子化学数据库的设计及应用应考虑分子与材料的通用性数据信息、以及含能材料领域重点关注的能量与稳定性的专用性数据信息,具有鲜明的特色性。对其做如下展望:(1)含能化合物的量子化学理论计算在方法选择、数据呈现方式上没有统一的标准。因此数据库的设计需要在计算基准方法与数据模型角度进行规定,并且随着计算方法的进步、更准确更有效方法的确认而不断更新;(2)数据库与高通量虚拟筛选相结合的含能化合物分子设计已有较多的研究报道,应用前景广阔;(3)相对于含能材料及其相关物性质的数据敏感性,含能化合物量子化学计算数据易于实现开放共享,是探索含能材料组成、结构与性能关系和设计新型含能材料的重要研究基础;(4)含能材料的量子化学计算包括分子以及晶体,相比于分子结构的量子化学计算,晶体结构的理论计算方法,有待系统的方法验证并制定基准方法。
