2D-LC-IT-TOF-MS/MS 用于国产阿莫西林颗粒聚合物杂质一致性评价
2023-11-03李佳蕊韩彬张雪霞任凤芝徐艳梅闫凯王巧高燕霞
李佳蕊,韩彬,张雪霞,任凤芝,徐艳梅,闫凯,王巧,高燕霞
1.河北省药品医疗器械检验研究院,河北 石家庄 050021;2.华北制药集团新药研究开发有限责任公司,河北 石家庄 050021;3.河北医科大学,河北 石家庄 050017
阿莫西林是口服半合成β-内酰胺类抗生素,抗菌谱广,是儿童及成人院外感染性疾病常用药[1],但其高发的过敏反应,尤其是过敏性休克,严重危害人民生命健康,需引起高度重视。已有研究[2-4]得知,阿莫西林过敏反应主要由聚合物引发。另有文献[5]报道,口服阿莫西林制剂聚合物含量与制剂中水分等工艺属性呈正相关。因此,聚合物杂质的控制,是阿莫西林颗粒关键质量属性,然而该品种现行质量标准《中国药典》2020 年版二部,未对该类杂质进行控制。
参考文献[6]方法,同时结合《中国药典》2020年版阿莫西林原料有关物质测定法[7],建立C18 色谱柱-高效液相色谱法测定6 批国产制剂样品及2批原研制剂样品中聚合物杂质,同时分离出的杂质用二维液相-在线脱盐-质谱联用仪进行结构确认。
本实验采用二维液相-阀切换-在线脱盐-质谱联用技术(2D-LC-IT-TOF-MS/MS )对聚合物杂质进行鉴定[8-9],该方法不改变原流动相条件,通过阀切换功能,弃去不能进入质谱的流动相组分,实现了目标杂质精准定位的液质联用分析[10],其中1stD-LC(一维液相)使用《中国药典》该品种有关物质项下色谱条件进行分析,通过共用的组分收集环捕集目标杂质;在1stD-LC 分析结束后,收集环中的杂质依次进行2ndD-LC(二维液相)分析,其中2ndD-LC色谱柱条件是适合进入质谱分析的流动相[9-12]。
1 仪器与试药
1.1 仪器
本实验使用岛津2D-LC-IT-TOF-MS/MS 系统。具体配置为LC-20AD 输液泵,SIL-20AC 自动进样器,CTO-20AC(柱温箱),CBM-20A(系统控制器),DGU-20A5(在线脱气机),SPD-M20A(二极管阵列检测器),SPD-20A(UV 检测器),FCV-12AH×3(流路选择阀),FCV-14AH×2(色谱柱切换阀),LCMS-IT-TOF(离子阱-飞行时间质谱仪)和LC MS solution Ver.3.70(色谱工作站)。
1.2 试药
阿莫西林系统适用性对照品(批号130608—201804,购买自中国食品药品检定研究院)。阿莫西林颗粒(规格:0.25 g,批号:F8082901、F8082902、F8 082903;规格:0.125 g,批号:F9022901、F9022902、F9 022903;厂家:河北某厂家)。原研样品(规格:250 mg/5 mL,批号:536R;规格:125 mg/5 mL,批号:V78F;厂家:葛兰素史克公司)。
2 方法与结果
2.1 液相色谱条件
1stD-LC 色谱条件。色谱柱:Thermo Hypersil GOLD C18(250 mm×4.6 mm,5 μm);检测波长:230 nm;柱温:35 ℃;流速:1.0 mL/min;进样量:20 µL;流动相A:0.05 mol/L 磷酸盐缓冲液(取0.05 mol/L 磷酸二氢钾溶液,用2 mol/L 氢氧化钾溶液调节pH 值至5.0)-乙腈(99∶1);流动相B:0.05 mol/L 磷酸盐缓冲液(pH 5.0)-乙腈(80∶20);先以流动相A-流动相B(92∶8)等度洗脱,待阿莫西林峰洗脱完毕后立即按表1 线性梯度洗脱[13]。

表1 梯度洗脱程序
2ndD-LC 色谱条件。色谱柱:InertSustainTMC18(2.1 mm×50 mm,2 μm);检测波长:230 nm;柱温:35 ℃;流速:0.3 mL/min;进样量:20 µL;自动进样器温度:4 ℃;流动相:水-乙腈(70∶30)等度洗脱。
2.2 质谱条件
ESI(+)和ESI(-)离子源;接口电压1.5 kV;雾化气:氮气1.5 L/min;干燥气:氮气10 L/min;碰撞气:氩气;脱溶剂管温度:150 ℃;加热模块温度:150 ℃;检测器电压:1.6 kV;一级质谱扫描范围:m/z200~3 000;二级质谱扫描范围:m/z200~1 500;校准方法:自动调谐,外标法校准质量数。
2.3 分析用样品溶液配制
精密称取阿莫西林系统适用性对照品,用流动相A 溶解并定量稀释成2 mg/mL 溶液,作为阿莫西林系统适用性对照品溶液;阿莫西林颗粒活性成分为阿莫西林三水合物,为了得到更多的阿莫西林聚合物,取过效期样品约2.5 g,置10 mL 量瓶中,加pH 6.0 磷酸二氢钾缓冲液溶解,室温放置72 h 后,作为阿莫西林聚合物混合溶液[14]。
2.4 1stD-LC 色谱分析
精密量取阿莫西林系统适用性对照品溶液和阿莫西林聚合物混合溶液各20 μL,分别注入高效液相色谱-质谱联用仪,记录色谱图。
阿莫西林系统适用性对照品溶液色谱图如图2所示,阿莫西林主峰、已知二聚体和三聚体的出峰时间,分别为6.637 min、28.516 min 和32.509 min。
阿莫西林聚合物混合溶液在该色谱系统中出峰如图3 所示,阿莫西林主峰出峰时间为6.625 min,与图1 一致,故推测28.623 min 处为阿莫西林二聚体,32.575 min 处为阿莫西林三聚体,并且检出的杂质峰中存在阿莫西林更高聚合体的可能。

图1 2D-LC-IT-TOF-MS/MS 杂质鉴定系统流路图

图2 阿莫西林系统适用性对照品溶液色谱图

图3 阿莫西林聚合物混合溶液色谱图
LOOP 环捕集保留时间分别为6.625 min(阿莫西林主峰),28.623 min(未知目标杂质峰F1),32.575 min(未知目标杂质峰F2),35.480 min(未知目标杂质峰F3),37.797 min(未知目标杂质峰F4),38.397 min(未知目标杂质峰F5)的目标杂质峰,用于LC/MS-IT-TOF 测定。
2.5 阿莫西林主峰高分辨质谱
阿莫西林分子式为C16H19N3O5S,应用分子量计算软件得理论分子量为365.104 5,质谱图见图4,其中m/z366.102 9 为[M+H]+,与计算分子量一致。

图4 阿莫西林主峰质谱图
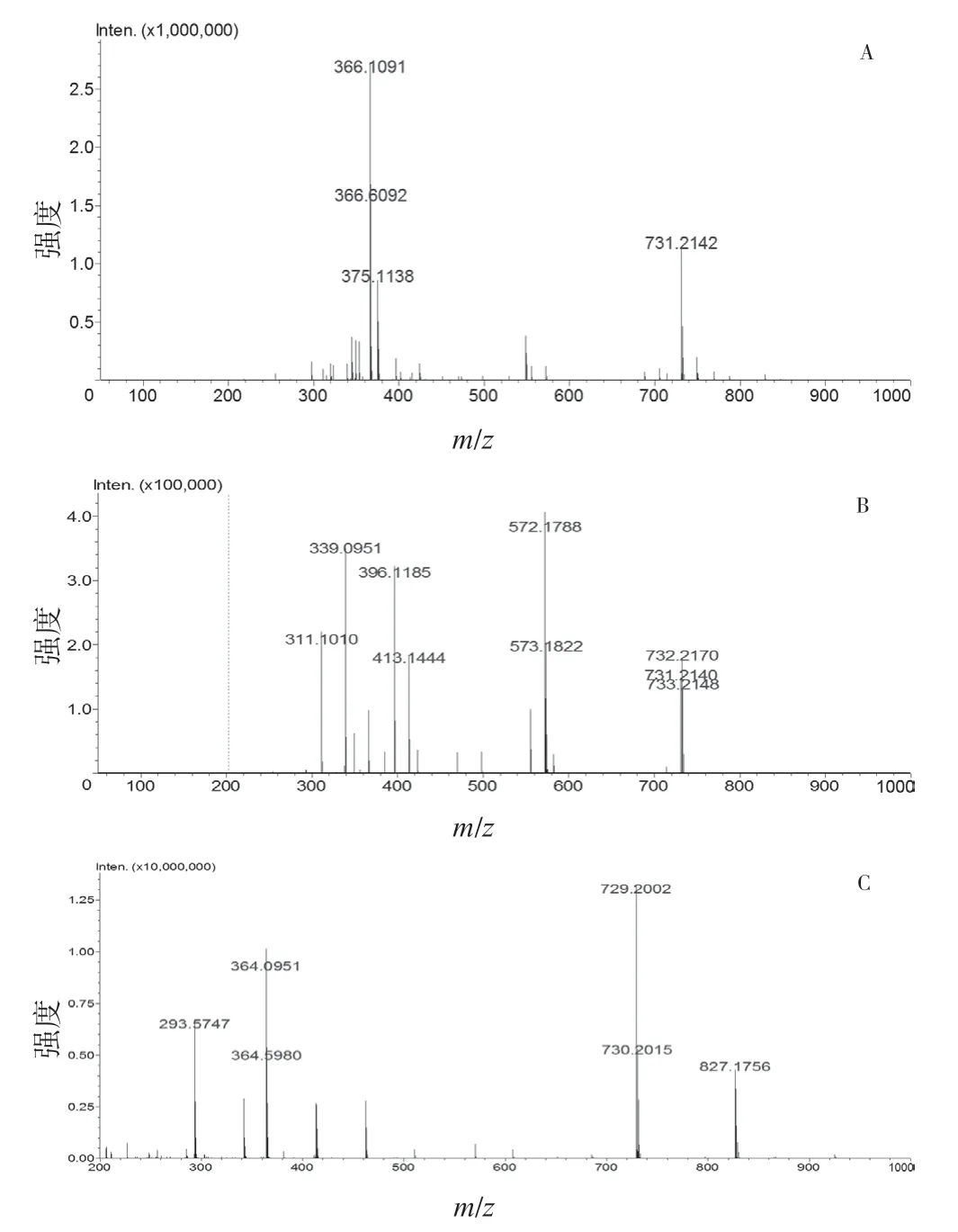
图5 未知目标杂质峰F1多级质谱图:A.正离子模式一级质谱图;B.正离子模式二级质谱图;C.负离子模式一级质谱图
2.6 未知目标杂质峰F1(28.623 min)
一级质谱图中m/z731.214 2 的离子推测为[M+H]+,说明目标峰相对分子量为730.214 2,是阿莫西林理论分子量2 倍,m/z366.109 1 碎片推测为聚合位点的酰胺键断裂形成的阿莫西林单体碎片。在二级质谱图中存在m/z572.178 8 和m/z413.144 4碎片离子,说明分子中存在2个159 Da的青霉素母核。
负离子模式一级质谱图中m/z729.200 2 的离子推测为[M-H]-,与正离子模式一致,因此,推测28.623 min 处未知目标杂质峰为阿莫西林二聚体。
2.7 未知目标杂质峰F2(32.575 min)
一级质谱图中m/z1 096.285 3、m/z1 134.268 7的离子,推测分别为[M+H]+和[M+K]+,说明目标峰相对分子量为1 095.313 8,是阿莫西林理论分子量3 倍,而m/z732.263 3 碎片推测为聚合位点的酰胺键断裂形成的阿莫西林二聚体碎片,见图6A。在二级质谱图中存在m/z937.289 3、m/z778.244 5 和m/z619.211 1 碎片离子,说明分子中存在3 个159 Da 的青霉素母核,见图6B。

图6 未知目标杂质峰F2多级质谱图:A.正离子模式一级质谱图;B.正离子模式二级质谱图;C.负离子模式一级质谱图
负离子模式一级质谱图中m/z1 094.297 5 的离子推测为[M-H]-,与正离子模式一致,因此,推测32.575 min 处未知目标杂质峰为阿莫西林三聚体,见图6C。
2.8 未知目标杂质峰F3(35.480 min)
一级质谱图中m/z1 461.612 5、m/z1 499.579 2的离子,推测分别为[M+H]+和[M+K]+,说明目标峰相对分子量为1 460.612 5,是阿莫西林理论分子量4 倍,而m/z1 096.286 3 碎片推测为聚合位点的酰胺键断裂形成的阿莫西林三聚体碎片(图7A)。在二级质谱图中存在m/z1 461.785 2、m/z1 302.477 1、m/z 1 144.357 3 和m/z985.311 4 碎片离子,说明分子中存在4 个159 Da 的青霉素母核(图7B)。

图7 未知目标杂质峰F3多级质谱图:A.正离子模式一级质谱图;B.正离子模式二级质谱图;C.负离子模式一级质谱图
负离子模式一级质谱图中m/z 1 459.430 5 的离子推测为[M-H]-,与正离子模式一致,因此,推测35.480 min 处未知目标杂质峰为阿莫西林四聚体(图7C)。
2.9 未知目标杂质峰F4(37.797 min)
一级质谱图中m/z1 826.554 4 的离子推测为[M+H]+,说明目标峰相对分子量为1 825.554 4,是阿莫西林理论分子量5 倍(图8A)。在二级质谱图中存在m/z1 667.472 7、m/z1 508.452 7、m/z1 349.413 6、m/z 1 190.387 6 和m/z1 031.340 5 碎片离子,说明分子中存在5 个159 Da 的青霉素母核(图8B)。

图8 未知目标杂质峰F4多级质谱图:A.正离子模式一级质谱图;B.正离子模式二级质谱图;C.负离子模式一级质谱图
负离子模式一级质谱图中m/z1 824.489 2 的离子推测为[M-H]-,与正离子模式一致,因此,推测37.797 min 处未知目标杂质峰为阿莫西林五聚体(图8C)。
2.10 未知目标杂质峰F5(38.397 min)
一级质谱图中m/z2 191.581 9 的离子,推测为[M+H]+,说明目标峰相对分子量为2 190.581 9,是阿莫西林理论分子量6 倍(图9A)。在二级质谱图中存在m/z1 096.281 8 碎片离子,为阿莫西林三聚体碎片(图9B)。

图9 未知目标杂质峰F5质谱图:A.正离子模式一级质谱图;B.正离子模式二级质谱图;C.负离子模式一级质谱图
负离子模式一级质谱图中m/z2 189.563 2 的离子推测为[M-H]-,与正离子模式一致,因此,推测38.397 min 处未知目标杂质峰为阿莫西林六聚体(图9C)。
经上述分析结果表明,阿莫西林可产生的聚合物杂质——阿莫西林二聚体、阿莫西林三聚体、阿莫西林四聚体、阿莫西林五聚体、阿莫西林六聚体,在测试用色谱条件下,均可得到有效分离。
3 样品测定
分别取6 批样品,2 批原研样品,按照“2.1”项下液相条件进样,结果见图10~12,有关物质测定结果见表2。

表2 有关物质测定结果 %

图10 国产制剂有关物质色谱图(批号:F8082901)
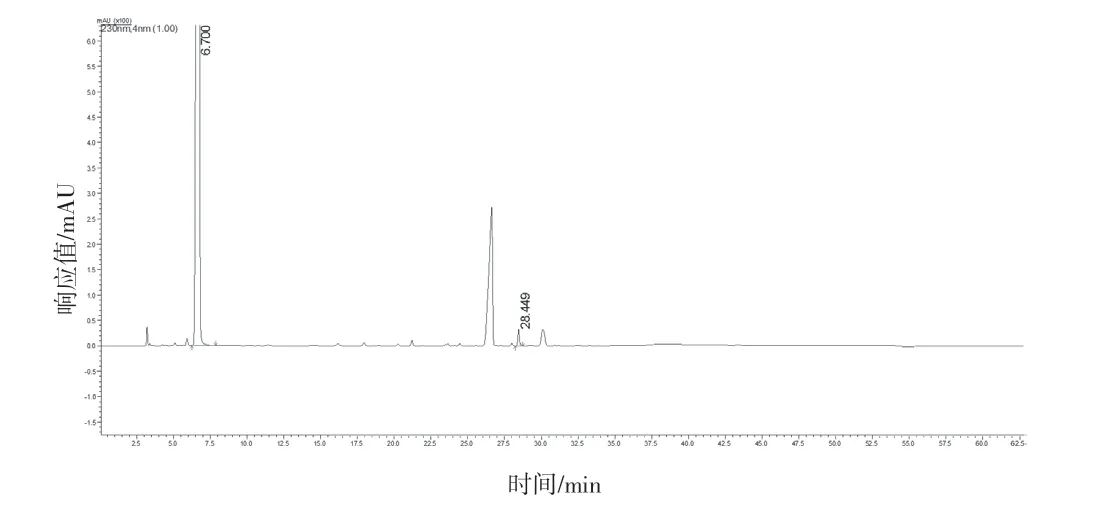
图11 原研样品有关物质色谱图(批号:V78F)

图12 阿莫西林对照溶液色谱图
4 结论
本研究建立的高聚物测定方法灵敏、准确,可在检测有关物质的同时进行聚合物的控制,其中阿莫西林四聚体、五聚体和六聚体为首次检出。
对比《中国药典》2020 年版二部收载的阿莫西林原料有关物质检查方法中254 nm 检测波长,阿莫西林各杂质在230 nm 处有更强的吸收,提高了检测的灵敏度;同时,细化梯度洗脱条件,改善了杂质检出量和分离效果,测定结果更加准确、真实体现产品纯度。
按照建立的方法,考察自研制剂与原研样品中聚合物杂质含量,企业内控标准中增加聚合物检查项的样品,产品中杂质检出数量及含量均显著降低,且批次间稳定。可见,全面考察产品质量,将质量标准与生产工艺紧密结合,准确寻找关键质量属性,可及时排除风险,有效提高产品质量,同时确保产品质量的持续一致性。
目前,我国药品消费市场主体仍是仿制药,通过质量一致性评价,促进生产工艺调整、改进,进而提高药品质量[15],对保证用药安全性和质量可控性都具有重要意义。
