转录后选择性多聚腺苷酸化调控与糖尿病肾病
2023-11-02赵婷婷综述刘志红审校
赵婷婷 综述 刘志红 审校
[作者单位]东部战区总医院 国家肾脏疾病临床医学研究中心(南京,210016)
选择性多聚腺苷酸化(alternative polyadenylation,APA)是真核细胞 mRNA 成熟过程中针对pre-mRNA的一种加工修饰方式,主要发生在3′UTR区,其通过选择不同的多聚腺苷酸化信号位点(polyadenylation signal site,PAS),产生编码序列相同但 3′UTR 序列长度不同的 mRNA 亚型。APA主要通过改变 3′UTR序列长度影响对应的反式作用因子(miRNA 或RNA结合蛋白等)的调控作用,进而调节 mRNA定位、稳定性、翻译效率及蛋白定位。本文将从APA的概况、检测方法、生物学功能、基因遗传和单细胞层面的最新研究进展、以及糖尿病肾病APA的研究现状和前景等方面进行阐述。
APA概况
APA作为重要的转录后调控机制,是在pre-mRNA 3′端序列中的顺式调控元件、反式作用因子,例如多种酶和蛋白因子等共同作用下完成,同时受多种细胞微环境因素的影响[1-2]。APA形成过程包括PAS下游切割位点的选择和多聚腺苷酸化两个重要环节,其中PAS 定位是APA形成的关键。人类约70%的基因含有多个PAS基序,分别称为近端PAS(或称为非经典的PAS)和远端PAS(或称为经典的PAS),经典的PAS基序是AAUAAA,非经典的PAS基序变体有AUUAAA、AGUAAA 和 UAUAAA 等,切割位点位于PAS下游10~30个核苷酸区域。此外,APA 的形成还需要其他高度保守的顺式元件参与,如 PAS 上游的 UGUA 序列和下游的 U-/GU-rich 序列,以上保守序列通过募集特定蛋白复合物共同调控APA的形成,包含CPSF、CSTF、CFI 和 CFII 等[3]。总之,在APA形成的过程中,CPSF复合物能准确识别PAS基序,同时CSTF和CFI复合物分别与PAS下游U-/GU-富集序列和上游UGUA模体等保守基序特异性结合,最终CPSF、CFI 和 CSTF 等复合物形成一个整体,协同完成pre-mRNA的PAS下游位点精确切割[2]。最后,在多聚腺苷酸聚合酶的催化作用下,在切割位点处添加多聚腺苷酸尾(图1)[1]。不同PAS位点的选择导致3′UTR序列长度不同,近端PAS上游的3′UTR序列称为基本UTR序列,而将其下游对应的序列称为选择性UTR序列。PAS不同位点的选择接受APA调节因子的调控,其中NUDT21,CPSF6,CFI59,U1 snRNP,PABPC1,HuR等调节因子通过与近端PAS附近的顺式作用原件结合而竞争性抑制APA调节复合物与之结合,进而促进远端PAS位点的选择,最终导致基因3′UTR序列整体延长,而CstF64的高表达会促进近端PAS位点的选择,进而引起基因3′UTR序列的整体缩短[1-2]。

图1 APA调控的顺式作用原件和对应的反式作用因子[2]
APA检测方法
目前已有一些专门针对APA研究的检测方法,主要包括PAS-seq,3′RNA-seq和3′RACE等,其中PAS-seq和3′RNA-seq是直接进行多聚腺苷酸位点的高通量测序的方法,通过对转录本中基因组编码序列和多聚腺苷酸尾的连接序列进行测序,最终确定PAS切割位点[4-5]。而3′RACE实验则是基于逆转录PCR技术从样本中快速扩增目的基因3′端序列的方法,该方法提高了检测PAS位点精确位置的灵敏度[6]。除了以上APA直接高通量测序的方法,我们还可通过常规的 RNA-seq 数据借助生信算法定量分析APA的动态变化。目前常用的推算方法包括DaPars、QAPA、GETUTR和Roar等11种算法[2]。Chen等[7]研究者将以上算法进行比较发现DaPars和QAPA两种算法的假阳性率较低,得到的结果准确性较高。其中DaPars是一种denovo的算法,是在不考虑之前的任何关于 APA 注释数据库情况下,基于自己的数据从头推算APA的方法[8];而QAPA 算法是整合已经构建的关于PAS的数据库来分析APA的动态变化[9]。
APA生物学功能
APA通过选择不同的PAS位点导致3′UTR的序列长度不同,在近端PAS和远端PAS间的UTR区域常含有与miRNA或RNA结合蛋白(RNA binding proteins,RBPs)等相结合的特定序列。因此,APA主要是通过改变3′UTR 序列长度影响对应的反式作用因子的调控作用,进而调节mRNA定位、稳定性、翻译效率以及蛋白定位(图2)。目前研究报道选择性多聚腺苷酸化与多种生理、病理过程密切相关,但其调节方向和调控机制存在差异[10]。3′UTR变短的基因主要参与细胞周期调控、RNA剪切加工、DNA扩增、蛋白磷酸化等生物学功能,而3′UTR变长的基因主要富集在细胞外基质沉积、细胞黏附、补体激活等进程[11]。例如在以肿瘤为主的增殖性疾病中,Xia 等[8]研究发现在肺癌、子宫癌、乳腺癌和膀胱癌中存在APA的调节,其中91%基因3′UTR显著变短,导致3′UTR 序列上miRNA结合密度降低,从而减弱miRNA的抑制作用,提高翻译水平,最终促进肿瘤细胞的增殖和恶化。为了探究肿瘤细胞3′UTR变短的机制,Yang等[12]进行深入研究,他们首次发现了一类肿瘤特异性泛素连接酶MAGE-A11,MAGE-A11通过催化3′mRNA加工复合体的重要成员PCF11的泛素化和降解,驱使不同肿瘤类型的APA调控和3′UTR整体缩短,进而动态调节致癌基因和抑癌基因的表达。此外,PolH作为DNA聚合酶的Y家族成员,介导DNA转运合成,是DNA损伤耐受的主要调节蛋白[13]。Zhang等[14]发现用顺铂处理肿瘤细胞时,PoIH基因的3′UTR显著缩短,miRNA的抑制作用减弱,PoIH蛋白表达水平升高,说明在治疗过程中,肿瘤细胞可能通过APA机制促进PoIH蛋白表达,进而产生耐药,促进肿瘤进展。除肿瘤外,研究报道在心肌肥厚、肌营养不良、慢性淋巴细胞白血病患者基因的3′UTR序列同样存在普遍变短的现象[2]。

图2 APA调节生物学功能的作用模式[1]
而在细胞分化和发育的进程中,主要包括成肌细胞、胚胎干细胞和神经元的分化和发育,APA调控的基因倾向于选择远端PAS,导致APA调控基因的3′UTR序列整体延长,RBPs通过结合位于延长的3′UTR内的特殊线性序列基序或二级结构来调节mRNA亚细胞定位和蛋白翻译[15-16]。例如,An等[15]在小鼠海马神经元中发现两种3′UTR长度不同的BDNF亚型,短3′UTR BDNF亚型主要定位于胞体,而长3′UTR亚型在RNA结合蛋白的协助作用下靶向转运至树突而发挥特殊的生物学功能。同样,Berkovits等[17]研究表明CD47 mRNA存在两种长度不同的3′UTR APA亚型,短3′UTR CD47亚型定位于内质网,而长3′UTR亚型募集RNA结合蛋白HuR和SET4复合物至3′UTR区域,通过进一步激活RAC1促进CD47转位到细胞质膜行使功能。Cau教授团队进行了关于APA介导3′UTR的延长调节蛋白翻译的研究,他们发现在FGF9 mRNA 长3′UTR区包含调节翻译的UG-repeat序列,RNA结合蛋白FUBP3与该重复序列结合后在不影响mRNA半衰期的情况下增强FGF9蛋白的翻译[18]。
APA在基因遗传层面的研究
虽然APA发生在约70%的人类基因中,但目前缺乏APA与疾病风险和人类性状表型之间的广泛关联分析,此外,有关非编码SNP在不同人体组织中与APA及其表型特征和疾病的关系的研究也相对匮乏。因此,Li教授[19]团队开发了DaParsV2.0算法,此方法基于来自467个个体,46种组织的8 722套RNA-seq与匹配的全基因测序数据,共鉴定到大约40万个与APA相关的遗传变异位点并命名为APA数量性状位点(3′aQTL),这些3′aQTL关联的基因共有11 613个,占已注释基因的51%。通过遗传力估计分析作者发现3′aQTL能解释大约25.2%的APA变异和16.2%的基因表达变异。同时,作者首次构建了人类多组织APA遗传图谱并分析了3′aQTL在各个组织的分布特异性。研究者还进一步系统分析了3′aQTL与poly(A) 基序,RNA二级结构和 RBP结合位点的关联,发现3′aQTL可通过改变这些元件进而影响APA。最有意义的是,作者通过CLIP-seq发现了一个APA全新的调控因子LARP4,约30%的3′aQTL可以通过改变LARP4的结合位点来调节对应基因的APA。这为APA关联的遗传位点提供了一种全新分子调控机制,同时也为之后发现APA调控因子提供了一个新的角度。最后,Li等[19]还分析了3′aQTL与疾病的关系。通过关联分析23种常见人类疾病与表型相关SNP和3′aQTLs,鉴定到11.5%的组织特异的性状中富含3′aQTL变异。作者同时发现3′aQTLs与溃疡性结肠炎、原发性胆管炎和阿尔茨海默病等多种遗传相关的自身免疫性疾病存在关联。该项工作对解释人类复杂疾病风险易感位点有着重要的推动作用,为揭示人类复杂性状和疾病病因学提供了新的方向。
此外,Cui等[20]还构建了APA关联分析方法(3′aTWAS),用于鉴定APA关联的大脑疾病风险基因。作者基于17 300个RNA-seq数据及其匹配的基因组数据训练了包含人体49种组织的3′aTWAS预测模型,并应用此模型对11种大脑遗传疾病(包括帕金森综合征、阿尔茨海默症和渐冻症等)进行分析,最终鉴定出354个APA关联的大脑疾病风险基因,其中超过50%的基因不能被以前的TWAS方法找到。当然3′aTWAS预测模型也可被广泛的应用到其他人类复杂表型和疾病的研究中,从而鉴定APA关联的遗传疾病风险基因。
APA在单细胞层面的研究
同时,随着单细胞测序技术的飞速发展,研究者同时将APA的研究方向聚焦在单细胞水平,从单细胞层面揭示APA的细胞间异质性及不同组织、生物过程和疾病中不同细胞类型的APA的差异。其中Wang等[21]开发了单细胞多腺苷酸化测序方法,这是一种直接对转录物3′端进行测序的链特异性方法。他们通过这种测序方法分析了多种细胞系发现,在bulk数据中使用多个PAS位点的基因在每个细胞中只倾向于选择一个PAS位点。同时发现,在细胞同步化后,PAS位点变化的基因富集在细胞周期相关通路中,而差异表达的基因却没有富集到细胞周期的调控,进而从单细胞层面说明APA在细胞周期的调控中发挥重要作用。除了对3′端直接测序外,也可以通过算法从常规的单细胞测序数据中推算出每种细胞的PAS切割位点,目前常用的算法包括scSAAP,scDAPA,scAPAmod,scDaPars,scMAPA 和scAPAtrap 等[22]。借助以上算法研究者已成功构建了急性髓细胞白血病、非小细胞肺癌、分泌细胞分化和发育小鼠胚胎的单细胞APA全景图[23-26],揭示了细胞类型特异性的APA调节,进而在转录后APA水平上揭示了细胞的异质性,扩大了人们对细胞异质性的认知范围。因此,单细胞APA的发展不仅为我们研究不同或者罕见细胞类型的生物学调控提供了新思路,更有助于从单细胞层面理解基因表达调控的新机制。
APA在糖尿病肾病的研究进展和前景
目前关于APA在肾脏领域的研究相对比较局限,有少量研究报道在缺血再灌注损伤或单侧输尿管梗阻方法构建的急性肾损伤的小鼠模型中,APA介导近端小管NLRP3和FGF2基因的3′UTR序列变短,进而促进肾小管的炎症、凋亡和纤维化[27-28]。但关于APA是否在糖尿病肾病肾小球损伤中发挥作用及其调节方向的研究相对比较匮乏。我们中心前期首次通过借助DaPars和QAPA算法从50例糖尿病肾病和25例对照患者的整个肾小球RNA-seq测序数据中进行APA的分析[29],结果表明,95% APA 调节基因的3′UTR在糖尿病肾病患者肾小球中普遍延长,qRT-PCR实验证实糖尿病肾病患者以选择远端PAS切割位点为主。同时,促进远端 PAS 选择的 APA 调节因子 NUDT21、CPSF6、SNRNP70 和 PABPC1 的蛋白水平在糖尿病肾病患者组表达显著增强而在对照组表达较低。整合APA 推算结果和转录组-蛋白质组学数据发现,在所有 3′UTR 变长基因中,约31%基因在蛋白水平高表达,但仅有约 5%基因在 mRNA 水平高表达,说明 APA 介导的 3′UTR序列延长具有提高 mRNA 翻译效率的作用。随后,我们筛选3′UTR长度变化比较显著且在糖尿病肾病中发挥重要作用的CYB5R1基因进行验证。在高糖刺激的足细胞中分别过表达 CYB5R1 长、短 3′UTR 亚型,发现长 3′UTR 亚型的蛋白表达水平明显高于短 3′UTR 亚型。最后,结合 POSTAR2 数据库进一步分析发现,约80%基因通过 3′UTR 的延长至少增加一个RBPs结合位点,且蛋白翻译效率在 RBPs结合基因与非结合的基因间存在显著差异。因此,本课题组首次提出APA 作为糖尿病肾病新发现转录后调控机制,其主要通过延长mRNA 3′UTR 序列长度来增强对应 RBPs 的调控作用,进而提高蛋白翻译效率、促进糖尿病肾病的发生发展(图3)。此研究是关于APA在整个肾小球中的调节作用,但肾脏是一个多细胞器官,分为肾小球和肾小管间质,其中肾小球又包含足细胞、系膜细胞和内皮细胞等,肾小管间质包含近端小管、远端小管和集合管等细胞。但迄今为止,关于糖尿病肾病细胞类型特异性的APA调节尚无研究报道。
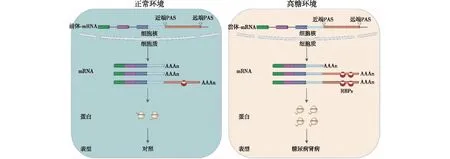
图3 糖尿病肾病肾小球APA调控的模式图[29]
与此同时,越来越多的研究者发现糖尿病患者个体间存在巨大差异,部分糖尿病患者在血糖控制良好的情况下发展为糖尿病肾病,而另一些患者在血糖控制欠佳的情况下仍保持正常的肾功能[30]。此外,糖尿病患者发生糖尿病肾病以及进展为终末期肾病具有家族聚集倾向,不同种族之间糖尿病患者发生糖尿病肾病的概率也不同[31],这些证据都提示遗传因素在糖尿病肾病发病中占有重要地位。目前,APA是否在糖尿病肾病的基因遗传层面发挥作用尚不清楚,以及与APA关联的遗传风险基因和易感位点仍是未知。
小结:随着高通量测序的飞速发展,不断涌现的糖尿病肾病全基因测序和单细胞测序数据为从基因遗传和单细胞层面研究糖尿病肾病APA的调控奠定了坚实的数据基础。因此,APA相关的遗传变异位点的筛选以及细胞特异的APA调节作用的研究将是今后糖尿病肾病APA研究的重点和方向。
