细胞分裂周期蛋白42通过内皮-间充质转化参与动脉型肺动脉高压小鼠右心室纤维化*
2023-11-02王晓彤秦立龙王寒黎汪丽静程玉生
王晓彤, 秦立龙, 王寒黎, 汪丽静, 程玉生△
(1皖南医学院第一附属医院心功能科,安徽 芜湖 241000;2皖南医学院第一附属医院呼吸与危重症医学科,安徽 芜湖 241000)
动脉型肺动脉高压(pulmonary arterial hypertension, PAH)是一种进行性、复杂性和毁灭性的疾病,由多种致病因素或遗传因素引起,最终导致右心衰竭和死亡[1-2]。由于发病机制尚未完全明了,目前现有的前列环素类似物、内皮素受体拮抗剂、磷酸二酯酶5 型抑制剂和可溶性鸟苷酸环化酶激动剂虽对PAH有一定疗效,但不甚理想,患者预后很差[3-4]。因此,阐明PAH右心纤维化发生发展的分子机制,寻找有效的干预靶点对PAH右心纤维化和功能不全的治疗具有重要意义。
内皮-间充质转化(endothelial-mesenchymal transition, EndMT)是内皮细胞失去部分细胞特征而获得间充质表型的过程,包括紧密连接缺失和细胞外基质生成增加,最初被观察到是在胚胎发育中心脏垫层形态心内膜细胞分化的一个生理过程,在心血管生物学的背景下,EndMT 在各种疾病中发挥作用,包括动脉粥样硬化、心脏瓣膜病、心脏纤维化和心肌梗死等[5-6]。
Rho GTP 酶是一种小蛋白(20~25 kD),属于Ras相关蛋白超家族,其中研究最多的是RhoA、Rac-1 和细胞分裂周期蛋白42(cell division cycle protein 42,Cdc42)。Cdc42在全身各组织中均有表达,参与细胞增殖、迁移、分化等生理过程。既往研究显示,角膜内皮细胞在成纤维细胞生长因子2 刺激下通过激活磷脂酰肌醇3-激酶导致EndMT,这一过程与Cdc42引起细胞肌动蛋白骨架改变有关[7]。RhoA 和Cdc42的缺失降低了α-平滑肌肌动蛋白(α-smooth muscle actin, α-SMA)的表达[8]。然而,Cdc42 是否通过调节EndMT 过程参与PAH 小鼠右心纤维化及重构,目前尚不明确。
本研究通过血管内皮生长因子受体2 抑制剂司马沙尼(semaxinib/SU5416)+缺氧(SU5416+hypoxia,SuHx)建立小鼠PAH 模型,探讨Cdc42 在PAH 小鼠右心纤维化中的作用及其机制研究。
材 料 和 方 法
1 实验动物及分组
18 只C57BL/6 雄性小鼠购于浙江维通利华实验动物技术有限公司,5~8 周龄,SPF 级,18~20 g,许可证号为SCXK(浙)2019-0001,伦理编号为2022236。将这18 只小鼠随机分成3 组:常氧对照(normoxia control, NC)组、PAH 模型(SuHx)组和SuHx+ML141(Cdc4抑制剂)组,每组6只。所有存活小鼠在4周后用异氟烷麻醉处死。
2 主要试剂与仪器
SU5416(HY-1034)购自MedChemExpress;ML141(S7686)购自Selleck;磁珠分选套装Mini & Midi-MACS Starting Kit(130-042-501)购自Miltenyi Biotec;内皮细胞培养液EGM-2(cc-3162)购自LONZA;α-SMA抗体(ab119952)和β-tubulin 抗体(ab6046)购自Abcam;β-actin 抗体(20526-1-AP)、Cdc42 抗体(10155-1-AP)和血小板内皮细胞黏附分子1(platelet endothelial cell adhesion molecule-1, PECAM-1/CD31)抗体(14395-1-AP)购自Proteintech;波形蛋白(vimentin)抗体(T55134)和血管内皮钙黏蛋白(vascular endothelial-cadherin, VE-cadherin)抗体(T56599)购自Abmart;;锌指蛋白Snail 抗体(3879S)购自Cell Signaling Technology。
Vevo 2100 超 声系统(VisualSonics);1.2F PV 3.5 mm导管和T400超声血流仪(Transonic)。
3 主要方法
3.1 小鼠PAH 模型建立和分组处理 NC 组为常氧4周;SuHx组[9]为每周腹腔注射SU5416(20 mg/kg)一次,同时放置于低氧(10% O2)箱4 周进行PAH 造模;SuHx+ML141 组是在SuHx 造模基础上,第2 周开始给予Cdc42 抑制剂ML141(8 mg/kg[10-11])腹腔注射,每天一次,直至4周。
3.2 右心室收缩压(right ventricular systolic pressure, RVSP)测定和经胸超声心动图 所有小鼠在4周后进行RVSP 和超声测定。使用脱毛膏将小鼠颈前及前胸部脱毛,暴露皮肤;装好架鼠板、连接温度传感和麻醉管;用3%的异氟烷将小鼠麻醉后,以1.5%的浓度维持麻醉,并固定于小动物超声仪加热板的动物四肢固定装置,维持动物体温,根据测定层面,调节动物体位。使用Vevo 2100 超声系统进行超声心动图,M-Mode 模式下以四腔心超声切面测量测三尖瓣环平面收缩期位移(tricuspid annular plane systolic excursion, TAPSE)。小鼠心脏彩超完成后在次日测定RVSP,所有小鼠均用1%异氟烷混合在100%氧气中,以1.0 L/min 的流速,使用麻醉机麻醉(期间注意观察小鼠状态,如呼吸频率,避免小鼠麻醉过度甚至死亡)。小鼠固定于洁净手术台上,通过右颈外静脉将Transonic 1.2F PV 3.5 mm 导管插入右心室。小动物生理仪AcqKnowledge 5.0 软件通过Transonic T400 心室压力容积及心输出量测量模块获得血流动力学信号,待信号稳定后收集血流动力学参数至少3 min,并进行RVSP测定。
3.3 细胞培养 通过磁珠分选试剂盒分选心脏内皮细胞,在内皮细胞培养液中培养2~3 代,每48 h 更新一次培养液。细胞实验分为三部分:(1)分为低氧诱导组(0、12、24、48 和72 h)和SuHx 组;(2)药物抑制组,设置浓度梯度(0、2、5 和10 µmol/L)ML141组[12-13];(3)分 为NC 组、低 氧72 h 组、低 氧72 h+ML141(10 µmol/L)组和NC+ML141(10 µmol/L)组。
3.4 心脏HE 和Masson 染色 取小鼠心脏,用4%多聚甲醛固定,石蜡包埋,切成5 µm 切片。为了确定心肌肥厚和心脏纤维化的程度,采用HE染色进行心肌染色,采用Masson 三色染色法对心脏胶原纤维化区域进行染色,并在Leica DM 2500-LED 光学显微镜下和Leica DF000T 相机2.5 倍物镜下进行检查。用ImageJ 软件(NIH)计算胶原容积分数(胶原面积/总面积)。
3.5 Western blot 已处理好的心脏组织和细胞样本置于配制好的裂解液(VRIPA∶VPMSF∶V蛋白酶抑制剂=50∶1∶1)中裂解。将收集好的细胞裂解物置于4 ℃预冷的离心机以12 000×g离心30 min。收集上清液,并使用Pierce BCA 蛋白质分析试剂盒测定蛋白质浓度。将含有等量蛋白质的裂解物与5× loading 缓冲液混合,100 ℃煮10 min 使蛋白变性。将蛋白上样于10% SDS 凝胶,进行电泳(80 V,130 min)分离,再电转(250 mA,180 min)移到0.22 µm PVDF 膜上。电转结束后将PVDF 膜置于封闭缓冲液(含5%脱脂牛奶的TBST,TBST 含0.1%吐温20)中室温孵育1 h。按实验目的不同选用Ⅰ抗(α-SMA、β-actin、Cdc42、vimentin、β-tubulin、VE-cadherin、CD31 和Snail 抗体,均1∶1 000)4 °C 孵育过夜。回收Ⅰ抗,用TBST 洗涤膜3 次,每次10 min。加入相应Ⅱ抗(1∶4 000),室温孵育1 h。孵育完成后,用TBST 洗膜3 次,每次10 min。向PVDF 膜加入已配制好的ECL 化学发光液,用Tannon全自动化学发光分析系统显像。用ImageJ分析软件对曝光好的条带进行灰度分析。
4 统计学处理
采用SPSS 18.0 软件进行统计学分析。数值均采用均数±标准差(mean±SD)表示。多组间比较采用单因素方差分析,组间两两比较采用最小显著性差异法(LSD法)。以P<0.05为差异有统计学意义。
结 果
1 SuHx组小鼠心脏组织Cdc42表达水平显著升高
Cdc42 在SuHx 组小鼠心脏组织中表达较NC 组显著升高(P<0.05),见图1。

Figure 1.The expression of Cdc42 was increased in mouse heart tissues of SU5416+hypoxia (SuHx) group.Mean±SD.n=5.*P<0.05 vs normoxia control (NC) group.图1 Cdc42蛋白在SuHx组和NC组小鼠心脏组织中的表达
2 Cdc42 抑制剂ML141 可降低SuHx 组小鼠RVSP并增大TAPSE
SuHx 组 小 鼠RVSP [(42.02±1.92) mmHg]较NC 组[(20.28±0.85) mmHg]显著升高(P<0.01),TAPSE [(0.67±0.06) mm]较NC 组[(1.03±0.08)mm]显著减小(P<0.01);与SuHx 组相比,SuHx+ML141 组小鼠RVSP [(34.59±1.60) mmHg]显著降低(P<0.01),而TAPSE [(0.86±0.14) mm]显著增大(P<0.01),见图2。

Figure 2.Administration of Cdc42 inhibitor ML141 decreased right ventricular systolic pressure (RVSP; A) and increased tricuspid annular plane systolic excursion (TAPSE; B) in SU5416+hypoxia (SuHx) group.Mean±SD.n=6.*P<0.05, **P<0.01 vs normoxia control (NC) group; ##P<0.01 vs SuHx group.图2 各组小鼠RVSP和心脏彩超TAPSE变化
3 Cdc421 抑制剂ML14 可减轻SuHx 组小鼠右心组织心肌细胞肥大及纤维化
右心组织HE 染色显示,SuHx 组较NC 组右室心肌细胞肥大、排列紊乱;右心组织Masson 染色显示,SuHx 组较NC 组右室(尤其是血管旁)纤维化显著增加(P<0.01);右心组织HE 和Masson 染色显示,SuHx+ML141组较SuHx组心肌细胞肥大和纤维化显著减轻(P<0.01),见图3。
4 SuHx组和低氧72 h组内皮细胞出现EndMT
SuHx 组和低氧(3% O2)72 h 组内皮细胞的End-MT 较正常内皮细胞(低氧0 h 组)明显增强,表现为EndMT 标志物CD31 和VE-cadherin 蛋白表达显著降低(P<0.05),vimentin 和α-SMA 蛋白表达显著升高(P<0.01),见图4。
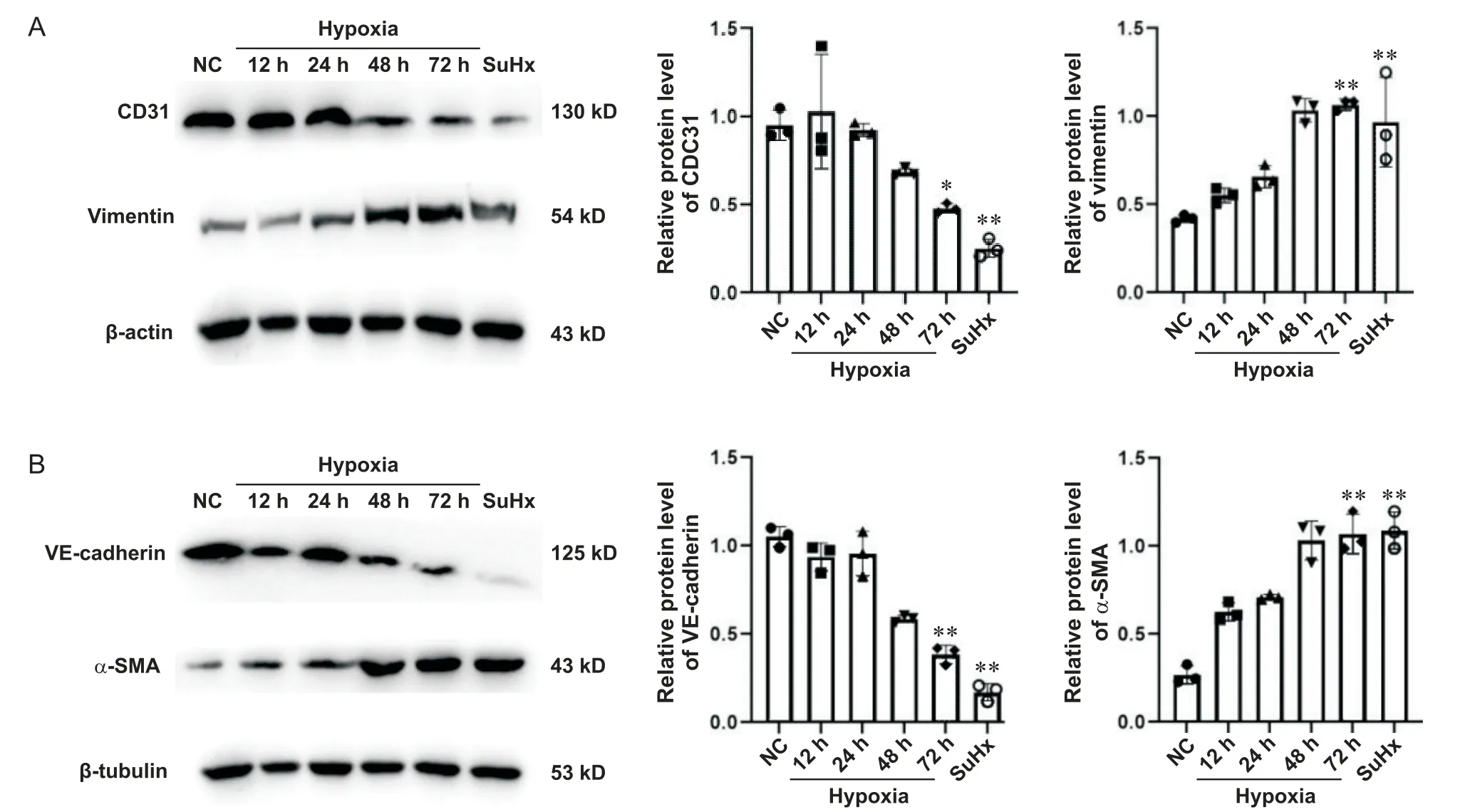
Figure 4.Endothelial-mesenchymal transition (EndMT) was observed in endothelial cells of SU5416+hypoxia (SuHx) group and hypoxia (72 h) group.A: the expression of EndMT-related proteins CD31 and viemntin in endothelial cells; B: the expression of EndMT-related proteins VE-cadherin and α-SMA in endothelial cells.Mean±SD.n=3.*P<0.05, **P<0.01 vs normoxia control (NC) group.图4 SuHx组和低氧72 h组内皮细胞出现EndMT
5 SuHx 组和低氧72 h 组内皮细胞Cdc42 蛋白表达均显著升高,抑制Cdc42可缓解内皮细胞EndMT
SuHx组和低氧72 h组内皮细胞Cdc42蛋白表达水平较正常内皮细胞(低氧0 h 组)显著升高(P<0.01),见图5A。低氧不同时点Cdc42的表达与End-MT 相一致。设置梯度浓度(0、2、5 和10 µmol/L)的Cdc42 抑制剂ML141 处理,结果显示在ML141 浓度为10 µmol/L 时缓解EndMT 作用最强,CD31 和VEcadherin 蛋白表达显著升高,vimentin 和α-SMA 蛋白表达显著降低(P<0.01),见图5B、C。

Figure 5.The increased expression of Cdc42 in endothelial cells of SU5416+hypoxia (SuHx) group and hypoxia (72 h) group, and the inhibitory effect of Cdc42 inhibitor ML141 on endothelial-mesenchymal transition (EndMT) induced by hypoxia for 72 h.A: the protein level of Cdc42; B: the expression of EndMT-related proteins CD31 and vimentin after treatment with different concentrations of ML141; C: the expression of EndMT-related proteins VE-cadherin and α-SMA after treatment with different concentrations of ML141.Mean±SD.n=3.**P<0.01 vs normoxia control (NC) group; #P<0.05, ##P<0.01 vs hypoxia (72 h) group; △P<0.05, △△P<0.01 vs 10 µmol/L ML141 group.图5 SuHx组和低氧72 h组内皮细胞Cdc42蛋白的高表达及ML141对EndMT的抑制作用
6 Cdc42抑制剂ML141改善内皮细胞EndMT形态
NC组内皮细胞呈铺路石样外观;SuHx组和低氧72 h 组内皮细胞出现EndMT,内皮细胞呈纺锤形外观;将预加了ML141 (10 µmol/L)的内皮细胞低氧诱导72 h(低氧72 h+ML141 组),纺锤形外观的内皮细胞较低氧72 h组减少,即EndMT被抑制,见图6。

Figure 6.Treatment with ML141 (10 µmol/L) reduced the spindle appearance of endothelial cells induced by EndMT in hypoxic (72 h) group(scale bar=200 µm).The endothelial cells in normoxia control(NC) group showed a paving stone-like appearance, while those in hypoxia (72 h) group and SU5416+hypoxia (SuHx) group showed a spindle appearance.图6 ML141对内皮细胞EndMT形态的影响
7 Cdc42抑制剂ML141抑制Snail蛋白表达
Snail 是EndMT 重要的转录蛋白。低氧72 h+ML141 组较低氧72 h 组的Snail 表达水平显著下降(P<0.05),见图7。

Figure 7.Treatment with ML141 (10 µmol/L) inhibited the expression of EndMT transcription protein Snail.Mean±SD.n=3.**P<0.01 vs normoxia control (NC) group; ##P<0.01 vs hypoxia (72 h) group.图7 ML141对内皮细胞EndMT转录蛋白影响
讨 论
PAH 最终导致右心纤维化和右心衰竭,心脏纤维化是许多终末期心血管疾病的共同病理表现和重要病理生理基础。如何改善右心功能纤维化与功能不全是一直致力研究的方向。本研究利用SuHx 构建PAH 模型,旨在探讨Cdc42 对PAH 右心纤维化的影响及其潜在机制。体内实验显示Cdc42在SuHx小鼠心脏组织中表达上调,抑制Cdc42 显著减轻PAH小鼠右心纤维化,改善右心功能。在体外实验中,Cdc42在低氧刺激的内皮细胞及SuHx内皮细胞表达上调,与EndMT 表达一致,给予Cdc42 抑制剂ML141处理后可以有效抑制EndMT。总之,这些数据表明Cdc42通过上调EndMT参与PAH后右心纤维化。
一般来说,用于PAH 新药物治疗临床前实验的常用动物模型有3 种:单氯萘啶肺损伤模型、慢性低氧性肺动脉高压模型和SuHx 模型。SuHx 模型可导致严重PAH,并与严重PAH 患者的血管变化非常相似,目前已被广泛使用。SU5416 作为一种血管内皮生长因子受体2 抑制剂,起初被用于肿瘤的治疗,后在PAH 研究中显示SU5416 产生的选择压力杀死了一些内皮细胞,并诱导常驻的内皮祖细胞转分化为平滑肌样细胞。与以往PAH 预防实验不同的是,SU5416 会导致肺血管内皮细胞初始凋亡,凋亡信号会刺激剩余细胞出现超增殖状态,并且在7 d内出现表型改变的内皮细胞[14],利用抗凋亡抑制剂可以缓解上述状态。本课题组前期在脂多糖模型中发现,肺血管内皮特异性敲除Cdc42可以抑制内皮细胞增殖和迁移,为避免早期SU5416 对内皮细胞凋亡信号的影响,本研究选择在SU5416 作用1 周后用Cdc42抑制剂ML141 进行实验。此外,SU5416 体外转分化的人肺微血管内皮细胞表明,EndMT 可能是PAH 复杂血管形成的机制[15],那么心脏血管内皮细胞是不是也存在EndMT?本研究显示,SuHx 组小鼠出现右心纤维化,并且SuHx 和低氧刺激心脏内皮细胞均出现EndMT,说明SuHx 模型中不仅肺血管内皮细胞出现EndMT,心脏血管内皮细胞也出现EndMT。炎症、低氧等情况会导致内皮细胞功能障碍,炎症介质如白细胞介素1β 和转化生长因子β(transforming growth factor-β, TGF-β)可诱导内皮细胞发生EndMT,病理性EndMT 参与压力超负荷、糖尿病和心肌梗死等诱导的心肌纤维化[16-18]。心脏内皮细胞出现EndMT 可能与SuHx 模型中低氧和炎症环境存在联系,EndMT引起组织纤维化可能跟基质金属蛋白酶有关。
Cdc42参与细胞的增殖、迁移、分化等生理过程,但大多数研究都是涉及与肿瘤相关的上皮-间充质转化(epithelial-mesenchymal transition, EMT),与调控EndMT相关的研究很少。我们的体内研究结果显示,Cdc42 抑制剂ML141 可以减轻右心心肌纤维化,减少胶原沉积,改善SuHx 小鼠右心功能。进一步体外研究结果显示,低氧刺激心脏内皮细胞后出现EndMT,而给予Cdc42 抑制剂ML141 可缓解EndMT,表明Cdc42可能通过调节EndMT参与PAH右心纤维化。Cdc42 是如何调控EndMT?缺氧和炎症参与PAH 致病过程,炎症因子TGF-β 可以激活Cdc42[19]。Cdc42 的关键靶标是一类肌动蛋白结合蛋白——肌动蛋白解聚因子/丝切蛋白(actin-depolymerizing factor/cofilin, ADF/cofilin)家族,该家族可以解聚肌动蛋白。Johnson 等[20]的报告表明,在表达和功能水平上,细胞骨架缺陷是骨形态发生蛋白受体II(bone morphogenetic protein receptor-II, BMPR-II)突 变 在BMPR-II 相关疾病肺动脉高压中的重要分子机制。BMPR-II 与LIM 结构域激酶1(LIM domain kinase 1,LIMK1)形成复合体(Cdc42 协同作用),通过ADF/cofilin 的磷酸化转换,将细胞外BMP 信号直接传递给肌动蛋白细胞骨架并导致细胞骨架重排[20-21]。本研究的显微镜观察结果显示,经历EndMT 的内皮细胞逐渐改变其致密和结构良好(所谓的鹅卵石样)的形状,并获得纺锤样形态。这种变化的部分原因是细胞肌动蛋白丝的细胞骨架重排,其根底取向的丧失,细胞连接的不稳定和细胞外基质的重塑。此外,Cdc42还可能通过调节内质网释放钙离子,改变线粒体膜电位及线粒体呼吸链复合体I、III 和V 活性,调节内皮细胞的通透性和迁移,来调控EndMT[22]。本研究未进一步验证Cdc42 通过何途径调控EndMT,但证明了Cdc42可以通过EndMT参与心脏纤维化。
EndMT 是一个复杂的过程,其进展包括广泛的中间表型和多个终点,会激活多种转录因子,最重要的是锌指转录因子Snail、Slug、Zeb1 和Zeb2,以及基本的螺旋-环-螺旋转录因子Twist-1,这些转录因子作为内皮细胞和间充质细胞基因表达的抑制因子和(或)激活因子,导致特征性内皮标志蛋白表达缺失,以及间充质蛋白表达增加[23-24]。低氧诱导EndMT 出现转录抑制因子Snail 的表达上调,Snail 直接促进内皮细胞向间充质表型分化。因此,增强的Snail 表达进一步验证了EndMT 的发生[25-26]。Snail 的上调不仅促进EndMT 产生大量成纤维细胞,还通过EndMT 细胞分泌的结缔组织生长因子诱导成纤维细胞向肌成纤维细胞转化,从而加速心脏纤维化的发展。Snail可能是心脏纤维化的潜在靶分子,本实验结果与既往实验结果相似。抑制Cdc42 可缓解EndMT 而导致Snail 表达水平降低,进一步说明Cdc42 可能是PAH右心纤维化的潜在治疗靶点。在癌症相关的EMT中,Snail 通常位于其他4 个因子的上游,但在EndMT中的研究表明,所有转录因子都可以强烈地影响彼此的表达。EndMT 的结果是否由于这些因素的共同作用,或者是否有一个主导因素,仍有待阐明。
EndMT 在心脏纤维化中的作用已被许多研究证实,成纤维细胞是在心脏纤维化等生理和病理条件下产生成细胞外基质的主要细胞类型。在一项开创性的研究中,Zeisberg 等[27]利用谱系追踪技术研究了实验性诱导的心脏纤维化成纤维细胞的起源,主动脉束带或同种异体移植排斥反应均可引起心脏纤维化,纤维化病变的定量分析显示,高达35%的成纤维细胞具有内皮细胞起源。但也有少数研究持不同观点,Moore-Morris 等[28]使用新型转基因示踪小鼠证明,在压力超负荷诱导的心脏纤维化期间,心脏成纤维细胞很少来源于内皮间充质转化细胞。其实在EndMT 病理过程中,内皮细胞出现EndMT 是复杂且受多种因素影响的过程。在与基因突变相关的疾病(如脑海绵状血管瘤)中,突变基因(过度)表达使内皮细胞发生EndMT。在这些情况下,内皮细胞的EndMT 能力可能由突变基因的表达水平决定。然而,在非遗传性疾病中,如细胞因子和生长因子的局部浓度、特定的细胞外基质结构或局部剪切应力条件,可能决定了一部分内皮细胞发生EndMT,但并非所有内皮细胞都是如此。另一方面,在谱系追踪实验中内皮标志物应仔细选择。首先,内皮细胞是一个表达不同标志物亚群的群体,这些标志物亚群有时会以时间和组织依赖的方式上调或下调。炎症或病理状态下的组织可能倾向于过度表达内皮细胞标志物,导致假阳性。因此,需使用一个以上标志物来验证。另外,基于患者来源细胞的新兴器官芯片平台将作为动物模型的补充来确定人类疾病过程中EndMT的具体机制。
目前我们的研究仅使用了Cdc42 抑制剂进行研究,后期可使用心脏内皮细胞Cdc42特异性敲除小鼠。同时本次实验样本量有限,未提取SuHx+ML141小鼠心脏内皮细胞进行实验比较,后期实验需增加样本量。我们也未对EndMT 具体信号通路进行研究,后期进行进一步研究是必要的。
综上所述,本研究证明了Cdc42 在体内和体外都是一种新型的EndMT 正调控因子。EndMT 在PAH 致右心纤维化及重塑的调控中起着重要作用,因此Cdc42 可能是治疗PAH 后心肌纤维化的一个有前景的靶点。
