大规模平行报告基因测定:一种分析基因表达调控的新技术
2023-10-30袁萌李辉王守志
袁萌,李辉,王守志
综 述
大规模平行报告基因测定:一种分析基因表达调控的新技术
袁萌1,2,3,李辉1,2,3,王守志1,2,3
1. 农业农村部鸡遗传育种重点实验室,哈尔滨 150030 2. 黑龙江省普通高等学校动物遗传育种与繁殖重点实验室,哈尔滨 150030 3. 东北农业大学动物科学技术学院,哈尔滨 150030
大规模平行报告基因测定(massively parallel reporter assay,MPRA)是一种可以同时研究基因组数千个调控元件活性的高通量分析方法。该方法在传统的荧光素酶报告基因载体上引入一段具有唯一标识的条形码,通过二代测序技术对转染前的DNA条形码和转染后的mRNA条形码进行测序,用mRNA和DNA条形码读数的比值来分析顺式调控元件的活性。自MPRA提出以来,已被广泛应用于基因组顺式调控元件和功能性变异的鉴定、转录后调控对表型的影响等方面的研究。本文对MPRA的发展历程、基本原理、实验流程、统计分析方法以及在顺式调控元件和转录后调控方面的应用进行了综述,并对其发展前景进行了展望,以期为相关领域研究人员了解与应用MPRA提供有益参考。
大规模平行报告基因测定;基因表达调控;顺式调控元件;转录后调控
基因表达调控一直是现代分子生物学基础研究的热点之一,其过程十分复杂,可分为染色质水平、转录水平、转录后水平、翻译水平和翻译后水平等调控[1,2]。转录和转录后水平上的调控是基因表达调控的重要部分,转录水平主要指特定顺式调控元件(cis-regulatory elements,CREs)与反式作用因子(trans-acting factors)相互作用而调控基因的表达。CREs是一段可调节基因表达的DNA序列,主要包括启动子、增强子和沉默子,它本身不编码任何蛋白质,但能够提供与反式作用因子相互作用的位点,进而参与基因表达调控[3,4]。因此,CREs的变异可能会通过改变转录因子结合位点来调控基因表达。转录后水平是指转录形成的前体信使核糖核酸(pre-mRNA)经过加工修饰成为信使RNA (mRNA)的过程,其主要包括RNA剪切、RNA编辑等[5~8]。pre-mRNA在加工修饰的过程发生突变不仅会影响RNA剪切[9]、RNA编辑[10]等过程,还可能影响mRNA稳定性[11],从而影响基因表达。目前,大多采用传统的双荧光素酶报告基因检测变异位点的调控活性,但每次只能检测单个变异位点,无法做到高通量检测[12~17]。
全基因组关联研究(genome-wide association study,GWAS)已被广泛用于检测与人类疾病和动植物经济性状相关的遗传变异的筛选,目前已经发现了大量的显著相关变异位点,但大多数变异位点位于基因组非编码区内,通过影响CREs来调控基因表达[18~20]。由于变异位点间存在强烈的连锁不平衡(linkage disequilibrium,LD),使得从众多变异位点中鉴别具有调控活性的变异位点(因果变异)具有很大的挑战性[21,22]。大规模平行报告基因测定(massively parallel reporter assay,MPRA)是解决这一难题的有效方法,该技术能够高通量检测GWAS关联信号区域中突变位点的等位基因活性差异,与基因编辑技术相结合能对功能性变异进行功能鉴定和验证,以鉴定可能的因果变异[23]。自MPRA提出以来,已广泛应用于人类疾病研究中,如鉴定与疾病相关调控元件的功能性变异,分析转录后调控对疾病的影响等[24~28]。本文对MPRA的发展历程、基本原理、实验流程、统计分析方法以及在CREs和转录后调控方面中的应用进行了总结,并对其发展前景进行了展望,以期为相关研究及其应用提供参考。
1 MPRA发展历程
从发展历程来看,MPRA技术发展可分为两类:第一类是基于有条形码的常规MPRA及其改进方法;第二类是基于无条形码的STARR-seq及其改进方法。表1总结了两类MPRA技术及其改进方法的目标序列来源、载体、转染方式、应用及其优缺点等信息。
2009年,Patwardhan等[29]首次提出一种以单核苷酸为研究对象对启动子进行高通量功能分析的方法,该方法需要大规模平行合成DNA序列并对其进行测序。在此基础上,Melnikov等[30]于2012年在对两种诱导型增强子(合成cAMP调节增强子和病毒诱导的干扰素β增强子)变异位点的研究中提出了MPRA这一技术。自此,随着MPRA技术的普及和发展,适用于各种实验的MPRA被不断的开发出来。2013年,Arnold等[31]开发了一种可以在全基因组范围内直接定量评估数百万候选增强子活性的技术——STARR-seq (self-transcribing active regulatory region sequencing)。与有条形码的MPRA相比,STARR-seq技术通过调控序列与启动子的相互作用来驱动报告基因和序列自身进行转录,调控序列的转录本作为标签而不需要条形码序列,使实验操作更为简便。在STARR-seq基础上,Vanhille等[32]于2015年开发了CapSTARR-seq技术,该技术克服了STARR-seq在哺乳动物中由于基因组过于复杂而使文库制备困难和测序深度过深的问题,为哺乳动物中增强子活性的研究提供了一种快速且经济的方法。至2016年为止,MPRA仅能在有限的细胞类型中检测短CREs的活性差异。为此,Shen等[33]开发了AAV MPRA (adeno-associated virus MPRA)技术以扩大MPRA的应用范围,该技术通过将DNA文库包装成AAV,使文库可以转导至广泛的组织中,从而能对任何可被AAV感染的组织或器官进行MPRA;同年,Inoue等[34]开发出一种基于慢病毒的MPRA技术,即lentiMPRA(lentivirus-based MPRA),该技术可用于任何被慢病毒有效感染的细胞,这使MPRA技术在生物学上的应用范围进一步扩大[35]。2018年,Kalita等[36]开发了一种检测调控序列内等位基因特异性表达的简化方法,称为BiT-STARR-seq (biallelic targeted STARR-seq)。该方法在反转录中引入了单分子标签(unique molecular identifiers,UMI),使得克隆和转化步骤中不会出现由于文库的复杂性而导致的误差,从而提高检测等位基因特异性表达的能力;同年,Wang等[37]提出了基于ATAC-seq (assay for transposase accessible chromatin with high-throughput sequencing)和STARR-seq的ATAC-STARR-seq技术,该技术可直接从开放染色质区域捕获目标片段,无需进行寡核苷酸合成,可以分析较长目标片段的活性。有研究表明,转录因子对DNA甲基化水平的敏感性普遍存在差异,但这种差异敏感性是否会转化为基因表达差异尚无定论[38]。因此,Lea等[38]于2018年开发了mSTARR-seq (methyl-STARR-seq)技术,主要用于研究DNA甲基化对数十万个基因片段(数百万个CpG位点)的调控作用,为分析DNA甲基化与基因表达水平之间的因果关系提供了有力工具。为了探索CREs和区域染色质是否以复杂的序列相互作用,以及它们与基因表达是否存在因果关系,Maricque等[39]于2018年开发了patchMPRA (parallel targeting of chromosome positions by MPRA)技术,该技术的关键点在报告基因产生的mRNA有两种不同的条形码:一个是用于指定CREs的CREs条形码(cBC);另一个是用于指定报告基因位置的基因组条形码(gBC),这使patchMPRA能够在不同的染色体位置测量同一组CREs,从而研究局部CREs和区域染色质对基因表达的调控作用。2023年,Zhao等[40]基于patchMPRA技术[39]的两种条形码模型开发了一种scMPRA(single-cell MPRA)技术,可检测具有细胞类型或细胞状态特异性的CREs,解决了传统的MPRA难以鉴定细胞类型或细胞状态特异性的CREs这一难题。由于在单细胞中回收mRNA的效率较低,因此可以从mRNA回收效率方面改进scMPRA。
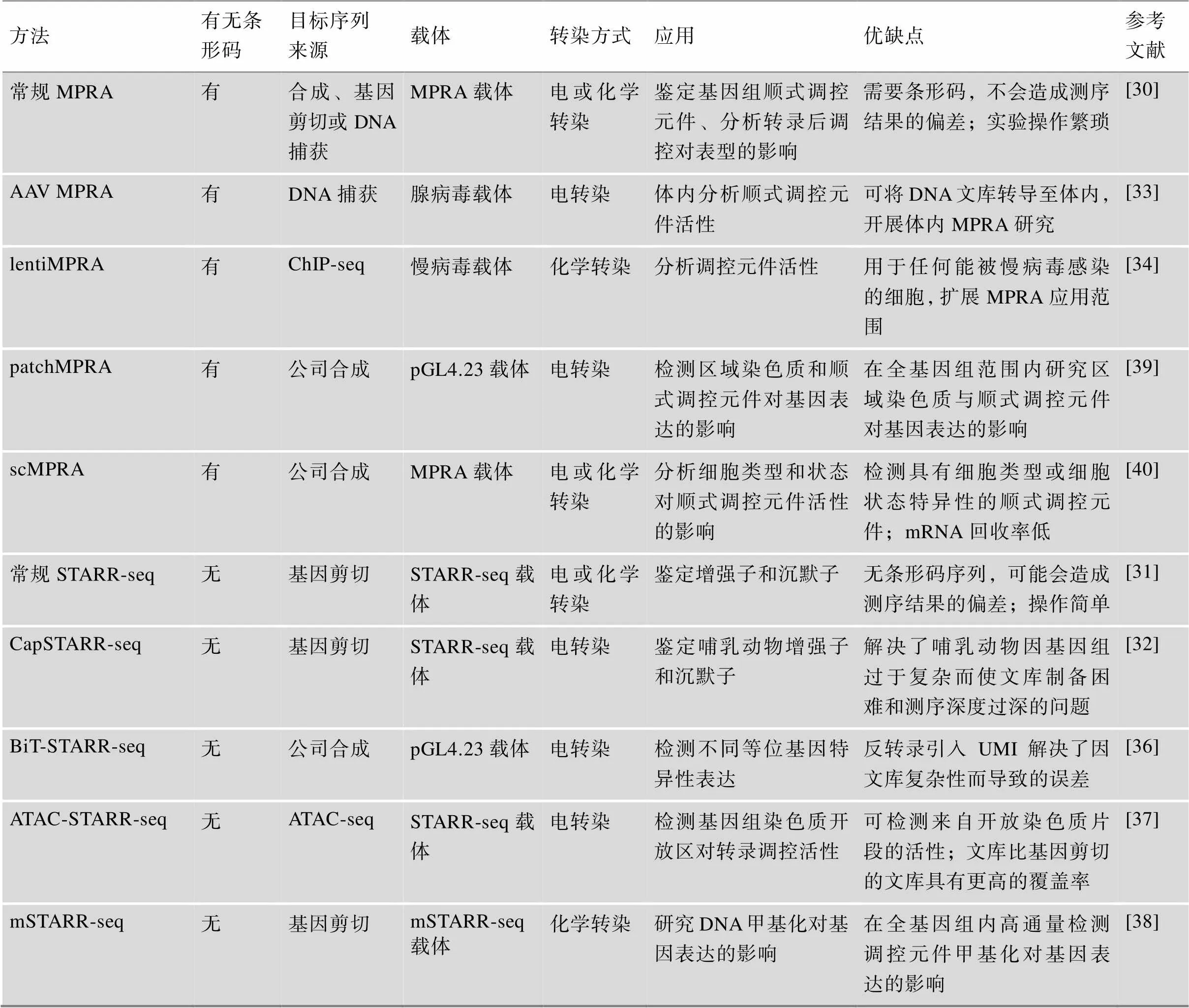
表1 不同MPRA技术的比较
2 MPRA基本原理、实验流程及统计分析
2.1 MPRA基本原理
简单来说,MPRA是一种升级版的荧光素酶报告基因分析法[41,42]。荧光素酶报告基因检测是一种以荧光素(luciferin)为底物检测萤火虫荧光素酶(firefly luciferase)活性的研究系统,萤火虫荧光素酶将荧光素催化氧化成氧化荧光素的过程中会产生生物荧光[43],可用于启动子结构和活性分析、miRNA与靶基因的靶向互作研究、转录因子与启动子调控机制研究以及其他信号转导通路研究等;方法是将特定的目标调控元件(增强子、启动子等)克隆到报告基因的载体中,构建成荧光素酶报告载体,然后瞬时转染至细胞中,培养48 h后回收细胞,用荧光测定仪(也称化学发光仪)测定荧光素酶活性[43]。有条形码的MPRA则是以荧光素酶报告基因为基础,在报告基因的3′端添加遗传条形码(一种随机寡核苷酸序列,可唯一标记匹配的目标序列),使得同一时间内可分析多个序列的调控活性[44]。一旦将构建好的DNA文库转染至细胞中,启动子就会驱动调控元件序列及其独特的条形码表达,通过二代测序技术量化条形码的转录效率来分析目标序列的调控活性,而非量化荧光素酶的发光[42]。STARR- seq技术在报告基因的3′端引入目标序列,有调控活性的目标序列作用于启动子使目标序列进行自转录,通过二代测序技术量化目标序列的转录效率以分析其调控作用[31]。就两种技术相比而言,STARR-seq与MPRA的主要区别是条形码的有无,STARR-seq不需要条形码,因此实验操作较简便,但该技术中报告基因与目标序列之间的稳定性可能会造成测序结果的偏差,而MPRA技术中的条形码可以很好的解决这一问题,但该技术实验操作较繁琐[35,45]。
2.2 MPRA实验流程
有条形码的MPRA实验流程主要由5部分构成:(1)构建DNA文库。在微阵列中合成目标DNA序列和条形码序列,将其连接后克隆至MPRA载体。之后,将通用启动子和开放阅读框(荧光素酶或绿色荧光蛋白等)克隆至目标DNA序列与条形码之间形成最终的DNA文库;(2)构建输入文库。通过二代测序技术对DNA文库中的条形码进行测序形成输入文库(DNA-seq);(3)转染。将DNA文库转染至细胞后,开放阅读框和条形码进行转录表达;(4)构建输出文库。转染一段时间后,收集细胞中的mRNA,对条形码进行测序形成输出文库(RNA-seq);(5)统计分析。整理RNA-seq和DNA-seq结果,通过它们读数的比值(RNA-seq/DNA-seq)反映目标DNA序列的相对表达量以分析其调控功能(图1A)。
与有条形码的MPRA相比,无条形码的STARR-seq技术的实验流程较为简便,主要由以下5部分组成:(1)构建DNA文库。通过基因组剪切等技术获得目标DNA片段,将其克隆至STARR-seq载体的开放阅读框(荧光素酶或绿色荧光蛋白等)和PloyA尾之间形成DNA文库;(2)构建输入文库。通过二代测序技术对DNA文库中的目标DNA序列进行测序形成输入文库(DNA-seq);(3)转染。将DNA文库转染至细胞中,有调控活性的目标DNA序列作用于启动子而促进该目标序列的自转录;(4)构建输出文库。转染一段时间后,收集细胞中的mRNA,对目标DNA序列的转录本进行测序形成输出文库(RNA-seq);(5)统计分析。整理RNA-seq和DNA-seq结果,通过它们读数的比值(RNA-seq/DNA-seq)反映目标DNA片段的相对表达量以分析其调控功能(图1B)。
A:常规MPRA实验流程;B:常规STARR-seq实验流程。
2.3 MPRA数据分析
MPRA是通过输出文库(RNA-seq)的mRNA与输入文库(DNA-seq)的DNA条形码或目标DNA序列读数的比值分析基因组中具有调控活性的序列和变异位点。最初,研究人员通常采用二项式检验[31]、差分峰值分析[46]、负二项式[47]及线性模型[48]等方法分析测序结果。随着MPRA的广泛应用,MPRA数据分析技术逐渐被开发出来,表2总结了相关分析方法的原理和优缺点。2017年,Kalita等[49]基于等位基因特异性定量分析(quantitative allele-specific analysis of reads,QuASAR)[50]开发了QuASAR- MPRA,该方法用β-二项式分布对RNA和DNA读数进行建模,提供了一种专门用于识别具有等位基因特异活性的调控序列的统计分析方法。QuASAR- MPRA虽然考虑了质粒比例的不平衡和测序误差等问题对测序结果的影响,简化了重复试验的统计分析,但不能通用于活性差异分析。为此,Myint等[51]于2019年开发出了一种通用于MPRA活性差异分析的方法,即mpralm。该方法基于voom方法使用线性模型进行活性差异分析[49],为需要复杂的数据分析模型的实验设计提供了灵活的分析方法。mpralm不仅功能全面,而且优于目前存在的分析方法。虽然MPRA实验技术越来越完善,但其数据分析还存在一定的不足,如:(1)忽略了数据中固有的噪音;(2)采用分析其他实验的数据模型;(3)现存专用于MPRA的分析方法只能用于活性差异分析,并且依赖于RNA与DNA比率的统计汇总,限制了实验数据的应用[48]。因此,Ashuach等[52]于2019年开发了MPRAnalyze分析框架,该方法使用图形模型将DNA和RNA读数联系起来,将数据直接建模为结构化数据。值得注意的是,MPRAnalyze内嵌套的广义线性模型使其能灵活应用于各种实验设计。有研究表明,一些为特定MPRA技术定制的分析方法,因MPRA实验设计的巨大差异、输入文件的复杂性等原因,无法在各种实验数据分析中相互转换应用[53]。为此,Gordon等[53]于2020年开发了一种基于Nextflow[54]的MPRA数据分析方法,即MPRAflow。该方法用DNA和mRNA测序结果拟合广义线性模型来分析每个被测CREs的转录效率。MPRAflow不仅可用Python、Bash和R语言自动运行MPRA处理代码,而且兼容了高性能计算(HPC)集群和云计算等多种计算体系,这些计算体系扩大了一些数据分析技术的应用范围[53]。同年,为解决由RNA二级结构、热力学稳定性和文库复杂性等对STARR-seq测序结果的影响,Lee等[45]开发了一个基于负二项式回归模型的分析方法,称之为STARRPeaker。该方法不仅可模拟基础转录效率,而且考虑了潜在的干扰因素。为了识别MPRA实验中的负调控元件(NRE),He等[55]于2022年设计了第一个适用于NRE识别的程序——Fast-NR,它通过整合测序读数和图形特征,利用STARR-seq实验产生的数据检测NRE。Fast-NR的应用将加速沉默子功能机制和表观遗传学研究。为解决数据质量和实验设计问题,Gilliot等[56]于2023年又开发了一个Python包,即FORECAST,该方法通过基于最大似然法的推断方法和Flow-seq实验概率模型对实验和分析参数进行系统的探索,有助于实验开发和数据的充分利用。

表2 MPRA数据分析技术的比较
3 MPRA在顺式调控元件及转录后调控中的应用
3.1 顺式调控元件
3.1.1 启动子
启动子是调控基因表达的顺式作用元件之一,它含有RNA聚合酶特异性结合和转录起始所需的保守序列,一般位于结构基因转录起始位点的上游,通过与转录因子结合而调控基因表达[57~59]。因此,启动子的突变可能会通过改变转录因子的结合位点而调控基因表达。Kircher等[60]采用饱和诱变MPRA分析与疾病相关的启动子功能性变异,在人肝癌细胞(HepG2)中进行两次生物学重复检测低密度脂蛋白受体基因(low-density lipoprotein receptor,)启动子的变异位点,在两次实验中均发现c.–152C>T和c.–142C>T突变使启动子活性降低,而c.–217C>T突变使其活性增强。此外,在人胚胎肾239T细胞(HEK293T)和胶质母细胞瘤中检测端粒逆转录酶基因(telomerase reverse transcriptase,)启动子的变异位点,发现c.–124C>T、c.–146C>T、c.–45G>T、c.–57A>C和c.–54C>A这5个变异位点均使启动子活性增强。Koesterich等[61]用LentiMPRA在神经干细胞中分析位于启动子区与孤独症谱系障碍(autism spectrum disorder,ASD)相关的3600个新生突变(variant,DNV)对基因表达的调控作用,经过MPRAflow和MPRAnalyze统计分析,发现487个野生型等位基因和473个突变型等位基因具有活性增强的趋势,165个变异为高置信度变异,但未发现ASD疾病中DNV富集的明显证据,也没有证据表明来自ASD的DNV在所有变异中都增强转录活性,这可能与细胞类型、DNV数量、测序深度等有关。这为今后研究非编码区DNV与神经发育障碍之间的因果关系提供了参考。Jores等[62]使用STARR-seq分析核心启动子的活性,选取18,329个拟南芥()、34,415个玉米()和27,094个高粱()核心启动子构成3种文库,分别克隆至有35S增强子和无35S增强子的两种载体中,分别转染至烟草()和玉米原生质体中,发现35S增强子可使核心启动子的活性增强,且核心启动子在烟草中具有更强的活性。此外,他们还发现双子叶植物拟南芥的启动子在双子叶植物烟草中活性更强,而单子叶植物玉米和高粱的启动子在单子叶植物玉米原生质体中表现出更强的活性。该研究揭示了不同类型的启动子与增强子之间的特定作用,为优化启动子提供了一种可行的实验方法。
3.1.2 增强子
增强子是一段位于非编码序列且具有增强基因表达功能的短DNA序列,其含有转录因子的结合位点,增强子通过与转录因子结合而提高靶基因的表达[63]。因此,增强子突变可能会通过影响转录因子结合位点调控基因表达。目前,MPRA已被广泛用于鉴定基因组中增强子序列以及其突变对基因表达的调控作用。Kalita等[36]通过BiT-STARR-seq对75,501个DNA片段进行活性差异分析,在人类淋巴母细胞系(lymphoblastoid cell line,LCL)中进行了9次生物学重复,通过QuASAR-MPRA分析鉴定出了2720个等位基因活性具有显著性差异的SNPs。该研究表明了BiT-STARR-seq这种简化的高通量报告基因检测方法分析基因组等位基因特异性表达的可行性,为今后的相关研究提供了一种新技术。Cooper等[23]用MPRA在HEK293T细胞中分析5706个非编码单核苷酸变异对基因转录活性的影响,这些变异位点来自阿尔茨海默病(Alzheimer’s disease,AD)和进行性核上性麻痹(progressive supranuclear palsy,PSP)的GWAS区域,共发现了320个具有调控活性的变异,随后利用CRISPR干扰或敲除鉴定并验证了多个风险位点。该研究表明了MPRA和CRISPR在高LD的复杂位点上分析与疾病相关的非编码区变异的有效性。Lu等[12]通过GWAS在系统性红斑狼疮(systemic lupus erythematosus,SLE)的91个风险位点中发现了3073个遗传变异,将其与先前发表的20个遗传变异构建了DNA文库,转染至由EB(epstein-barr virus)病毒感染的B淋巴细胞(GM12878)中,发现了482个变异具有增强子活性,其中51个变异在27个风险位点显示基因型依赖性增强子活性。为进一步探索具有细胞类型特异性的变异,将DNA文库转染至人T淋巴细胞瘤细胞(Jurkat)中,鉴定出92个等位基因变异,其中有25%变异在GM12878中被鉴定,这为今后的研究提供了宝贵的资源。该研究不仅为阐明SLE相关的转录调控机制提供了见解,而且为剖析人类复杂疾病的病因提供了蓝图。Wu等[64]用STARR-seq鉴定猪()基因组中的增强子序列,将滇南小耳猪基因组进行随机剪切构成DNA文库,转染至猪肾细胞(PK15)和猪睾丸细胞(ST)量化增强子的活性,在PK15和ST细胞分别检测到1015和2217个增强子,其中有601个增强子在两个细胞系中均存在,这表明STARR-seq可用于在全基因组范围内量化猪增强子的活性。该研究首次应用于猪的全基因组增强子活性量化图谱的绘制,它将促进猪复杂性状因果突变的鉴定。Sun等[65]用STARR-seq鉴定水稻(L.)基因组中的增强子序列,用从2周龄日本晴水稻幼苗茎上提取的基因组构建DNA文库,转染至原生质体中,在两次生物学重复试验中,分别发现了15,208和12,210个具有增强子活性的序列,其中9642个序列在两次重复中均显现出增强子活性。该研究通过功能分析定量鉴定水稻的增强子,为在不同生物学背景下进一步开展机理研究提供了宝贵资源。
3.1.3 沉默子
在遗传学中,沉默子是一段能够结合转录因子(阻遏蛋白)的DNA序列,与增强子对DNA转录的增强作用相反,沉默子会抑制DNA的转录过程[66]。DNA上的基因是信使RNA合成的模板,而信使RNA最终被翻译成蛋白质。当沉默子存在时,阻遏蛋白结合到沉默子DNA序列上,会阻碍RNA聚合酶转录DNA序列,从而阻碍RNA翻译为蛋白质[67]。Doni等[68]开发了SSA (simple subtractive analysis)算法以识别人类基因组内未鉴定的调控元件,用STARR-seq技术在K562细胞中分析7500个未鉴定的调控元件,发现41.5%具有沉默子活性。为进一步证实结果的可靠性,他们又用CRISPR-Cas9技术敲除鉴定的沉默子,发现其缺失使靶基因表达具有增强的趋势,该研究为全基因组沉默子的鉴定提出了一种通用策略,初步解决了计算或实验方法在全基因组范围内识别沉默子具有挑战性这一问题。Mouri等[69]通过MPRAduo在全基因组范围内分析神经元限制性沉默元件,用8436个有典型抑制元件1-沉默子转录因子(repressor element 1-silencing transcription factor,REST)结合位点的序列和4430个无典型REST结合位点的序列与5种增强子分别构成ES(增强子-沉默子)文库,将其转染至GM12878、HepG2、K562和人神经母细胞瘤细胞(SK-N-SH)中,发现2657个有典型REST结合位点的序列和486个无典型REST结合位点的序列表现出活性降低的趋势,该研究结果表明MPRAduo能够预测抑制元件1的活性及其变异对抑制元件1的影响,为转录因子结合位点的全基因组功能鉴定提供了一个范例。Hussain等[70]通过CapSTARR-seq技术检测小鼠()基因组内的沉默子,用小鼠双阳性胸腺细胞的28,055个DNA酶I超敏感位点(DHS)和437个非DHS区构建DNA文库,克隆至由3种不同启动子构成的STARR-seq载体,启动子为超核心启动子1 (SCP1)、人磷酸甘油酸激酶基因普遍存在的强启动子(pPGK)和T细胞特异性启动子增强子对pRag2-Eα(pR-Eα),将文库转染至来源于小鼠T细胞的P5424细胞系中,在SCP1、pPGK和pR-Eα文库分别检测出1249、672和413个沉默子,其中有15个沉默子在3种文库均被检测到。该研究将STARR-seq方法用作一种新的高通量分析策略,以定量评估哺乳动物中沉默子的活性,为全基因组沉默子的鉴定和特征描述提供了一种通用策略。
3.2 转录后调控
3.2.1 pre-mRNA剪接
在真核生物中,大多数的蛋白质编码基因都是转录为pre-mRNA后,通过加工去除非编码区(内含子)成为mRNA后再翻译成蛋白质[71]。剪接是pre-mRNA加工过程中的关键步骤,由一个超大蛋白质复合体(剪接体)催化,识别并作用于内含子与外显子连接处的特定序列(剪接位点),是一个动态且可调控的过程[72,73]。一般情况下,pre-mRNA含有多个假定剪接位点,其中的任何一个都可能是剪接体的对接位点。有相关研究表明,内含子和外显子的突变会通过引起剪接位点的改变而调控基因表达,进而引发疾病或经济性状差异[74~77]。随着高通量测序技术的发展,MPRA成为研究pre-mRNA剪接的一种日益流行的方法。Rosenberg等[9]用MPRA探究200多万个基因的可变剪接位点,他们将黄色荧光蛋白衍生物的序列分成两个外显子,并在两个外显子之间引入具有简并序列的内含子,每个内含子都设计两个相互竞争的剪接位点,并在两个相互竞争的位点之间或远端位点的下游位置插入两个长为25 bp的完全简并序列,由此创建了两个复杂性文库,一个是由265,137个序列组成的5′端剪接位点文库,另一个是由2,211,739个序列组成的3′端剪接位点文库。将文库转染至细胞中,通过机器学习用RNA-seq的丰富数据建立剪接模型,预测人类SNP对剪接的影响。值得注意的是,虽然MPRA不包括外显子跳跃文库,但该模型能预测外显子跳跃序列的变异。该研究提出了一个基于合成序列进行MPRA的框架,显著提高了对pre-mRNA剪接的理解和预测人类自然遗传变异影响的能力。Soemedi等[78]开发了一种大规模平行剪接测定(MaPSy)来筛选4964个外显子致病突变对剪接的影响。实验包括两方面,一个是通过将文库转染细胞评估突变对体内剪接的影响,另一个是在细胞核提取物中评估突变对体外剪接的影响。选择长度小于或等于100 bp含突变位点的外显子,合成为包含至少55 bp的上游内含子和至少15 bp的下游内含子的待测序列,根据测序获得等位基因读数,确定每对等位基因(突变型/野生型)的比率。虽然实验系统不同,但两种实验得到的结果一致,约有10%的外显子突变对剪接有影响。该研究首次提出一种对外显子剪接突变进行大规模分析的新型技术,为大规模鉴定和描述外显子剪接突变提供了便利。
3.2.2 RNA编辑
基因经过转录后和翻译后调控机制会产生不同的蛋白质,使真核生物产生不同的性状[79]。在转录后调控中,基因的pre-mRNA会经历各种加工修饰,如5′端加帽、剪接、3′端加尾等[80]。RNA编辑是转录后机制之一,它能通过编码基因组序列改变RNA序列,进而调控基因表达。RNA编辑主要发生在非编码区,少数发生在编码区[81,82]。RNA编辑除了能改变蛋白质序列外,还能影响剪接[83]、RNA稳定性[84]、翻译[85]等。相关研究表明,RNA编辑与多种疾病相关,如:癌症与肌萎缩侧索硬化症(amyotrophic lateral sclerosis,ALS)、帕金森病(Parkinson’s disease,PD)和AD等神经退行性疾病[81,86]。因此,鉴定功能性编辑位点不仅有助于了解主要生物学作用,而且有助于阐明RNA编辑与疾病的关系。目前,MPRA已经用于分析RNA编辑。Choudhury等[10]用MPRA筛选调控mRNA丰度的3′UTR功能性编辑位点,建立了一个包含770个位于3′UTR的A-to-G编辑位点的文库,通过比较位点在DNA文库和mRNA文库中未编辑和编辑的相对富集度,共发现214个编辑位点导致报告基因活性的显著变化,这表明相对较多的3′UTR编辑位点可能通过参与转录后调控机制调控基因表达。Safra等[27]采用MPRA验证假尿苷(Ψ)的位点,并分析导致其特异性的因素。该研究采用以Ψ为中心的65 bp序列和8 bp条形码的模式设计了6411个序列,将序列克隆至编码区(CDS)的下游形成文库,将文库转染至细胞后,用一种通过作用于Ψ而阻止转录的物质进行处理。因此,可通过高通量测序确定Ψ的确切碱基对位置,鉴定可导致尿苷转化为Ψ的DNA序列和变异,从而破解RNA编辑的大部分未知调控密码[42]。这为研究尿苷酸假尿苷化提供了一种高通量策略。
3.2.3 RNA稳定性
众所周知,储存在DNA中的遗传信息会通过mRNA传递给蛋白质[87],但该过程会受到多种水平上的调控,如:染色质调控、转录调控、转录后调控和翻译调控等。其中,转录后调控中的mRNA稳定性是影响蛋白质丰度的关键性因素,其受3′UTR和5′UTR影响[88,89]。有研究表明,MPRA可用于研究3′UTR和5′UTR序列与mRNA稳定性之间的关系[11,90,91]。Siegel等[92]基于fast-UTR(massively parallel functional annotation of sequences from 3′UTRs)技术分析人类3′UTR序列对mRNA稳定性的影响,将由41,288个3′UTR序列构建的文库转染至细胞中,发现mRNA稳定性随富含AU的元件(AU-rich element,ARE)的序列长度增加而显著降低。基于这一发现,他们提出一个新的ARE分类系统来分析ARE对基因表达和mRNA稳定性的影响,发现两次实验结果一致。通过进一步研究包括本构衰变元件(constitutive decay element,CDE)在内的其他基序,发现CDE茎环的长度对mRNA稳定性有显著影响,这表明该研究不仅可用于ARE,还可用于分析CDE。虽然该研究为解析人类3′UTR序列中的ARE与CDE提供了新策略,但其还有许多局限性,如:实验分析的是3′UTR的一个子集,而不是整个基因组;该策略主要是针对ARE和CDE而设计的,对于研究其他序列的可行性未知[92]。Jia等[91]通过MPRA分析由100多万个5′UTR变异组成的文库,文库转染至HEK293细胞中,不仅发现上游开放阅读框(uORF)和下游GFP之间的完全随机化的10 bp序列会导致mRNA翻译能力和稳定性的差异,而且还发现了一个位于5′UTR中非结构化的富含A的元件在没有翻译的情况下影响mRNA稳定性。该研究结果不仅揭示了5′UTR在控制mRNA可翻译性方面的多种序列特征,还揭示了依赖于核糖体和不依赖于核糖体的mRNA监控途径。
4 结语与展望
目前,MPRA已广泛应用于人类疾病或复杂性状的研究,但在动植物上的研究鲜有报道。尽管MPRA以其高通量的优势在人类疾病或复杂性状的基因组转录调控和转录后调控等研究中得到广泛的应用,并取得了显著的进展,但其还有一些局限性,主要体现在如下几个方面:(1)MPRA无法识别受调控元件影响的靶基因。MPRA需要与eQTL和Hi-C等功能基因组学技术结合使用以识别靶基因[42,93]。但当所用细胞为稀有细胞类型和遇到环境扰动时,获得与生物学背景相匹配的数据是具有挑战性的[42]。Perturb-seq能很好的解决这一问题,它针对MPRA验证的调控元件设计一个与条形码相关的gRNA文库,条形码能调控gRNA靶基因的表达[42]。将文库转染至可表达Cas9蛋白的细胞系中,gRNA扰乱感兴趣的区域,通过scRNA-seq分析转录组的数据变化,进而验证调控元件的功能[94],并揭示调控元件的靶基因以及相关通路。(2)MPRA在检测长目标序列受到限制。原因有两点,一个是寡核苷酸序列的合成能力有限,序列大多在300 bp以内,这使MPRA大多用于分析短目标序列的活性差异。虽然300 bp足够用于分析特定的转录因子结合位点、局部序列的相互作用等[35],但大部分完整的调控元件都大于300 bp。同时,相关研究表明MPRA结果受目标序列长度的影响最大,随着目标序列的增长,MPRA检测活性的能力也会增加[38,95]。Klein等[95]提出了一项用于合成长目标片段的新技术,即HMPA。未来,人们可以通过进一步的开发或改进HMPA等方法构建复杂且统一的长目标序列文库,扩大MPRA的应用领域和研究深度。另一个是STARR-seq虽然能通过随机剪切和捕获等方法来进行长目标序列的分析,但这些方法都需要大量的DNA样本[30,96]。因此,在应用此种方法研究稀有样本和濒临物种会面临巨大的挑战。(3)载体染色质的不确定性。AAV MPRA载体进入细胞后,AAV基因组会由单链DNA转为双链DNA,然后以环化单体或共聚体外显子的形式存在,从而获得染色质特性[97,98]。但人们不清楚衍生染色质是否接受了全套的修饰,不能确定载体是否能完全再现调控元件的活性特征[44]。lentiMPRA的载体整合到宿主基因组中会被所有表观遗传修饰因子标记,但整合位置效应对检测结果有影响[42]。将报告基因载体特异性插入至宿主基因组可以改善这一问题[99],但该方法还没有在MPRA中实现。(4)不同实验设计的一致性。随着MPRA的广泛应用,其实验策略越来越多样化,各种策略之间的一致性也成为研究者需要考虑的因素。Klein等[95]基于经典MPRA、STARR-seq和lentiMPRA设计了9种实验策略,并分析实验结果的一致性。结果表明,目标序列位于最小启动子5′UTR而条形码位于报告基因3′UTR的lentiMPRA、目标序列和条形码分别位于最小启动子和报告基因5′UTR的lentiMPRA、以ORI(the bacterial plasmid origin-of-replication)为启动子的STARR-seq和经典MPRA有良好的一致性,而与以HSS为启动子的STARR-seq、目标序列和条形码分别位于最小启动子和报告基因3′UTR的lentiMPRA没有良好的一致性。这表明实验策略之间存在差异,要求研究者在制定研究策略时要考虑好各方面的因素,避免引起不必要的误差。
MPRA不仅可以用于研究人类疾病,还能用于研究生物进化的问题。如,结合祖先序列重建法检测调控元件在进化过程中的活性变化[100];结合序列扫描法鉴定功能性等位基因并绘制精细图谱[35]等。总之,随着科学技术的不断发展,新的研究方法以及技术会不断出现,为基因组学研究带来巨大的发展空间。MPRA作为一种高通量、应用范围广的研究基因表达调控的利器,未来在人类疾病、动植物重要性状的遗传机理解析等领域将得到更广泛的应用。
[1] Venters BJ, Pugh BF. How eukaryotic genes are transcribed., 2009, 44(2–3): 117–141.
[2] Mercer TR, Dinger ME, Mattick JS. Long non-coding RNAs: insights into functions., 2009, 10(3): 155–159.
[3] Di Iulio J, Bartha I, Wong EHM, Yu HC, Lavrenko V, Yang DC, Jung I, Hicks MA, Shah N, Kirkness EF, Fabani MM, Biggs WH, Ren B, Venter JC, Telenti A. The human noncoding genome defined by genetic diversity., 2018, 50(3): 333–337.
[4] Nord AS, West AE. Neurobiological functions of transcriptional enhancers., 2020, 23(1): 5–14.
[5] Newman A. RNA splicing., 1998, 8(25): R903–R905.
[6] Nimmich ML, Heidelberg LS, Fisher JL. RNA editing of the GABA(A) receptor alpha3 subunit alters the functional properties of recombinant receptors., 2009, 63(4): 288–293.
[7] Shi YG. Mechanistic insights into precursor messenger RNA splicing by the spliceosome., 2017, 18(11): 655–670.
[8] Corbett AH. Post-transcriptional regulation of gene expression and human disease., 2018, 52: 96–104.
[9] Rosenberg AB, Patwardhan RP, Shendure J, Seelig G. Learning the sequence determinants of alternative splicing from millions of random sequences., 2015, 163(3): 698–711.
[10] Choudhury M, Fu T, Amoah K, Jun HI, Chan TW, Park S, Walker DW, Bahn JH, Xiao XS. Widespread RNA hypoediting in schizophrenia and its relevance to mitochondrial function., 2023, 9(14): eade9997.
[11] Rabani M, Pieper L, Chew GL, Schier AF. A massively parallel reporter assay of 3′UTR sequences identifies in vivo rules for mRNA degradation., 2017, 68(6): 1083–1094.e5.
[12] Lu XM, Chen XT, Forney C, Donmez O, Miller D, Parameswaran S, Hong T, Huang YB, Pujato M, Cazares T, Miraldi ER, Ray JP, de Boer CG, Harley JB, Weirauch MT, Kottyan LC. Global discovery of lupus genetic risk variant allelic enhancer activity., 2021, 12(1): 1611.
[13] Abell NS, DeGorter MK, Gloudemans MJ, Greenwald E, Smith KS, He ZH, Montgomery SB. Multiple causal variants underlie genetic associations in humans., 2022, 375(6586): 1247–1254.
[14] Wang WJ, Li YD, Li ZW, Wang N, Xiao F, Gao HH, Guo HS, Li H, Wang SZ. Polymorphisms of KLF3 gene coding region and identification of their functionality for abdominal fat in chickens., 2021, 7(3): 792–799.
[15] Li Z, Liu X, Li Y, Wang W, Wang N, Xiao F, Gao H, Guo H, Li H, Wang S. Chicken C/EBPζ gene: expression profiles, association analysis, and identification of functional variants for abdominal fat., 2021, 76: 106631.
[16] Chatterjee S, Ahituv N. Gene regulatory elements, major drivers of human disease., 2017, 18: 45–63.
[17] Cheng BH, Zhang H, Liu C, Chen X, Chen YF, Sun YH, Leng L, Li YM, Luan P, Li H. Functional intronic variant in the retinoblastoma 1 gene underlies broiler chicken adiposity by altering nuclear factor-kB and SRY-related HMG box protein 2 binding sites., 2019, 67(35): 9727–9737.
[18] Araujo AC, Carneiro PLS, Alvarenga AB, Oliveira HR, Miller SP, Retallick K, Brito LF. Haplotype-based single-step GWAS for yearling temperament in American Angus cattle., 2021, 13(1): 17.
[19] Welter D, MacArthur J, Morales J, Burdett T, Hall P, Junkins H, Klemm A, Flicek P, Manolio T, Hindorff L, Parkinson H. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations., 2014, 42(Database issue): D1001–D1006.
[20] Barrett LW, Fletcher S, Wilton SD. Regulation of eukaryotic gene expression by the untranslated gene regions and other non-coding elements., 2012, 69(21): 3613–3634.
[21] Tewhey R, Kotliar D, Park DS, Liu B, Winnicki S, Reilly SK, Andersen KG, Mikkelsen TS, Lander ES, Schaffner SF, Sabeti PC. Direct identification of hundreds of expression-modulating variants using a multiplexed reporter assay., 2016, 165(6): 1519–1529.
[22] Ulirsch JC, Nandakumar SK, Wang L, Giani FC, Zhang XL, Rogov P, Melnikov A, McDonel P, Do R, Mikkelsen TS, Sankaran VG. Systematic functional dissection of common genetic variation affecting red blood cell traits., 2016, 165(6): 1530–1545.
[23] Cooper YA, Teyssier N, Dräger NM, Guo QY, Davis JE, Sattler SM, Yang ZA, Patel A, Wu S, Kosuri S, Coppola G, Kampmann M, Geschwind DH. Functional regulatory variants implicate distinct transcriptional networks in dementia., 2022, 377(6608): eabi8654.
[24] Choi J, Zhang TW, Vu A, Ablain J, Makowski MM, Colli LM, Xu M, Hennessey RC, Yin JH, Rothschild H, Gräwe C, Kovacs MA, Funderburk KM, Brossard M, Taylor J, Pasaniuc B, Chari R, Chanock SJ, Hoggart CJ, Demenais F, Barrett JH, Law MH, Iles MM, Yu K, Vermeulen M, Zon LI, Brown KM. Massively parallel reporter assays of melanoma risk variants identify MX2 as a gene promoting melanoma., 2020, 11(1): 2718.
[25] Sample PJ, Wang B, Reid DW, Presnyak V, McFadyen IJ, Morris DR, Seelig G. Human 5′UTR design and variant effect prediction from a massively parallel translation assay., 2019, 37(7): 803–809.
[26] Mulvey B, Dougherty JD. Transcriptional-regulatory convergence across functional MDD risk variants identified by massively parallel reporter assays., 2021, 11(1): 403.
[27] Safra M, Nir R, Farouq D, Slutskin IV, Schwartz S. TRUB1 is the predominant pseudouridine synthase acting on mammalian mRNA via a predictable and conserved code., 2017, 27(3): 393–406.
[28] Rhine CL, Neil C, Wang J, Maguire S, Buerer L, Salomon M, Meremikwu IC, Kim J, Strande NT, Fairbrother WG. Massively parallel reporter assays discover de novo exonic splicing mutants in paralogs of Autism genes., 2022, 18(1): e1009884.
[29] Patwardhan RP, Lee C, Litvin O, Young DL, Pe’er D, Shendure J. High-resolution analysis of DNA regulatory elements by synthetic saturation mutagenesis., 2009, 27(12): 1173–1175.
[30] Melnikov A, Murugan A, Zhang XL, Tesileanu T, Wang L, Rogov P, Feizi S, Gnirke A, Callan CG, Kinney JB, Kellis M, Lander ES, Mikkelsen TS. Systematic dissection and optimization of inducible enhancers in human cells using a massively parallel reporter assay., 2012, 30(3): 271–277.
[31] Arnold CD, Gerlach D, Stelzer C, Boryń ŁM, Rath M, Stark A. Genome-wide quantitative enhancer activity maps identified by STARR-seq., 2013, 339(6123): 1074–1077.
[32] Vanhille L, Griffon A, Maqbool MA, Zacarias-Cabeza J, Dao LTM, Fernandez N, Ballester B, Andrau JC, Spicuglia S. High-throughput and quantitative assessment of enhancer activity in mammals by CapStarr-seq., 2015, 6: 6905.
[33] Shen SQ, Myers CA, Hughes AEO, Byrne LC, Flannery JG, Corbo JC. Massively parallel cis-regulatory analysis in the mammalian central nervous system., 2016, 26(2): 238–255.
[34] Inoue F, Kircher M, Martin B, Cooper GM, Witten DM, McManus MT, Ahituv N, Shendure J. A systematic comparison reveals substantial differences in chromosomal versus episomal encoding of enhancer activity., 2017, 27(1): 38–52.
[35] Romero IG, Lea AJ. Leveraging massively parallel reporter assays for evolutionary questions., 2023, 24(1): 26.
[36] Kalita CA, Brown CD, Freiman A, Isherwood J, Wen XQ, Pique-Regi R, Luca F. High-throughput characterization of genetic effects on DNA-protein binding and gene transcription., 2018, 28(11): 1701–1708.aa
[37] Wang XC, He L, Goggin SM, Saadat A, Wang L, Sinnott-Armstrong N, Claussnitzer M, Kellis M. High-resolution genome-wide functional dissection of transcriptional regulatory regions and nucleotides in human., 2018, 9(1): 5380.
[38] Lea AJ, Vockley CM, Johnston RA, Del Carpio CA, Barreiro LB, Reddy TE, Tung J. Genome-wide quantification of the effects of DNA methylation on human gene regulation., 2018, 7: e37513.
[39] Maricque BB, Chaudhari HG, Cohen BA. A massively parallel reporter assay dissects the influence of chromatin structure on cis-regulatory activity., 2019, 37: 90–95.
[40] Zhao SQ, Hong CKY, Myers CA, Granas DM, White MA, Corbo JC, Cohen BA. A single-cell massively parallel reporter assay detects cell-type-specific gene regulation., 2023, 55(2): 346–354.
[41] Liu S, Liu YW, Zhang Q, Wu JY, Liang JB, Yu S, Wei GH, White KP, Wang XY. Systematic identification of regulatory variants associated with cancer risk., 2017, 18(1): 194.
[42] McAfee JC, Bell JL, Krupa O, Matoba N, Stein JL, Won H. Focus on your locus with a massively parallel reporter assay., 2022, 14(1): 50.
[43] Jiwaji M, Daly R, Pansare K, McLean P, Yang JL, Kolch W, Pitt AR. The Renilla luciferase gene as a reference gene for normalization of gene expression in transiently transfected cells., 2010, 11: 103.
[44] Zheng YJ, VanDusen NJ. Massively parallel reporter assays for high-throughput in vivo analysis of cis-regulatory elements., 2023, 10(4): 144.
[45] Lee D, Shi MM, Moran J, Wall M, Zhang J, Liu J, Fitzgerald D, Kyono Y, Ma LJ, White KP, Gerstein M. STARRPeaker: uniform processing and accurate identification of STARR-seq active regions., 2020, 21(1): 298.
[46] Muerdter F, Boryń ŁM, Woodfin AR, Neumayr C, Rath M, Zabidi MA, Pagani M, Haberle V, Kazmar T, Catarino RR, Schernhuber K, Arnold CD, Stark A. Resolving systematic errors in widely used enhancer activity assays in human cells., 2018, 15(2): 141–149.
[47] Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2., 2014, 15(12): 550.
[48] Law CW, Chen YS, Shi W, Smyth GK. Voom: precision weights unlock linear model analysis tools for RNA-seq read counts., 2014, 15(2): R29.
[49] Kalita CA, Moyerbrailean GA, Brown C, Wen XQ, Luca F, Pique-Regi R. QuASAR-MPRA: accurate allele-specific analysis for massively parallel reporter assays., 2018, 34(5): 787–794.
[50] Harvey CT, Moyerbrailean GA, Davis GO, Wen XQ, Luca F, Pique-Regi R. QuASAR: quantitative allele-specific analysis of reads., 2015, 31(8): 1235–1242.
[51] Myint L, Avramopoulos DG, Goff LA, Hansen KD. Linear models enable powerful differential activity analysis in massively parallel reporter assays., 2019, 20(1): 209.
[52] Ashuach T, Fischer DS, Kreimer A, Ahituv N, Theis FJ, Yosef N. MPRAnalyze: statistical framework for massively parallel reporter assays., 2019, 20(1): 183.
[53] Gordon MG, Inoue F, Martin B, Schubach M, Agarwal V, Whalen S, Feng SY, Zhao JJ, Ashuach T, Ziffra R, Kreimer A, Georgakopoulos-Soares I, Yosef N, Ye CJ, Pollard KS, Shendure J, Kircher M, Ahituv N. lentiMPRA and MPRAflow for high-throughput functional characterization of gene regulatory elements., 2020, 15(8): 2387–2412.
[54] Di Tommaso P, Chatzou M, Floden EW, Barja PP, Palumbo E, Notredame C. Nextflow enables reproducible computational workflows., 2017, 35(4): 316–319.
[55] He N, Wang WJ, Fang C, Tan YJ, Li L, Hou CH. Integration of count difference and curve similarity in negative regulatory element detection., 2022, 13: 818344.
[56] Gilliot PA, Gorochowski TE. Effective design and inference for cell sorting and sequencing based massively parallel reporter assays., 2023, 39(5): btad277.
[57] Slobodin B, Agami R. Transcription initiation determines its end., 2015, 57(2): 205–206.
[58] Haberle V, Lenhard B. Promoter architectures and developmental gene regulation., 2016, 57: 11–23.
[59] Lai HY, Zhang ZY, Su ZD, Su W, Ding H, Chen W, Lin H. iProEP: a computational predictor for predicting promoter., 2019, 17: 337–346.
[60] Kircher M, Xiong CL, Martin B, Schubach M, Inoue F, Bell RJA, Costello JF, Shendure J, Ahituv N. Saturation mutagenesis of twenty disease-associated regulatory elements at single base-pair resolution., 2019, 10(1): 3583.
[61] Koesterich J, An JY, Inoue F, Sohota A, Ahituv N, Sanders SJ, Kreimer A. Characterization of de novo promoter variants in autism spectrum disorder with massively parallel reporter assays., 2023, 24(4): 3509.
[62] Jores T, Tonnies J, Wrightsman T, Buckler ES, Cuperus JT, Fields S, Queitsch C. Synthetic promoter designs enabled by a comprehensive analysis of plant core promoters., 2021, 7(6): 842–855.
[63] Arrowsmith CH, Bountra C, Fish PV, Lee K, Schapira M. Epigenetic protein families: a new frontier for drug discovery., 2012, 11(5): 384–400.
[64] Wu YQ, Zhang YD, Liu H, Gao Y, Liu YY, Chen L, Liu L, Irwin DM, Hou CH, Zhou ZY, Zhang YP. Genome-wide identification of functional enhancers and their potential roles in pig breeding., 2022, 13(1): 75.
[65] Sun JL, He N, Niu LJ, Huang YZ, Shen W, Zhang YD, Li L, Hou CH. Global quantitative mapping of enhancers in rice by STARR-seq., 2019, 17(2): 140–153.
[66] Gilbert W, Müller-Hill B. Isolation of the lac repressor., 1966, 56(6): 1891–1898.
[67] Ogbourne S, Antalis TM. Transcriptional control and the role of silencers in transcriptional regulation in eukaryotes., 1998, 331(Pt 1): 1–14.
[68] Doni Jayavelu N, Jajodia A, Mishra A, Hawkins RD. Candidate silencer elements for the human and mouse genomes., 2020, 11(1): 1061.
[69] Mouri K, Dewey HB, Castro R, Berenzy D, Kales S, Tewhey R. Whole-genome functional characterization of RE1 silencers using a modified massively parallel reporter assay., 2022, 3(1): 100234.
[70] Hussain S, Sadouni N, van Essen D, Dao LTM, Ferré Q, Charbonnier G, Torres M, Gallardo F, Lecellier CH, Sexton T, Saccani S, Spicuglia S. Short tandem repeats are important contributors to silencer elements in T cells., 2023, 51(10): 4845–4866.
[71] Maniatis T, Reed R. An extensive network of coupling among gene expression machines., 2002, 416(6880): 499–506.
[72] Kornblihtt AR, Schor IE, Alló M, Dujardin G, Petrillo E, Muñoz MJ. Alternative splicing: a pivotal step between eukaryotic transcription and translation., 2013, 14(3): 153–165.
[73] Irimia M, Blencowe BJ. Alternative splicing: decoding an expansive regulatory layer., 2012, 24(3): 323–332.
[74] Snyman M, Xu S. The effects of mutations on gene expression and alternative splicing.2023, 290(2002): 20230565.
[75] Qi T, Wu Y, Fang HL, Zhang FT, Liu SY, Zeng J, Yang J. Genetic control of RNA splicing and its distinct role in complex trait variation., 2022, 54(9): 1355–1363.
[76] Fabo T, Khavari P. Functional characterization of human genomic variation linked to polygenic diseases., 2023, 39(6): 462–490.
[77] French JD, Edwards SL. The role of noncoding variants in heritable disease., 2020, 36(11): 880–891.
[78] Soemedi R, Cygan KJ, Rhine CL, Wang J, Bulacan C, Yang J, Bayrak-Toydemir P, McDonald J, Fairbrother WG. Pathogenic variants that alter protein code often disrupt splicing., 2017, 49(6): 848–855.
[79] Iwanami Y, Brown GM. Methylated bases of ribosomal ribonucleic acid from HeLa cells., 1968, 126(1): 8–15.
[80] Cantara WA, Crain PF, Rozenski J, McCloskey JA, Harris KA, Zhang XN, Vendeix FAP, Fabris D, Agris PF. The RNA modification database, RNAMDB: 2011 update., 2011, 39(Database issue): D195–D201.
[81] Christofi T, Zaravinos A. RNA editing in the forefront of epitranscriptomics and human health., 2019, 17(1): 319.
[82] Walkley CR, Li JB. Rewriting the transcriptome: adenosine-to-inosine RNA editing by ADARs., 2017, 18(1): 205.
[83] Hsiao YHE, Bahn JH, Yang Y, Lin XZ, Tran S, Yang EW, Quinones-Valdez G, Xiao XS. RNA editing in nascent RNA affects pre-mRNA splicing., 2018, 28(6): 812–823.
[84] Brümmer A, Yang Y, Chan TW, Xiao XS. Structure- mediated modulation of mRNA abundance by A-to-I editing., 2017, 8(1): 1255.
[85] Bazak L, Haviv A, Barak M, Jacob-Hirsch J, Deng P, Zhang R, Isaacs FJ, Rechavi G, Li JB, Eisenberg E, Levanon EY. A-to-I RNA editing occurs at over a hundred million genomic sites, located in a majority of human genes., 2014, 24(3): 365–376.
[86] Krestel H, Meier JC. RNA editing and retrotransposons in neurology., 2018, 11: 163.
[87] CRICK FH. On protein synthesis., 1958, 12: 138–163.
[88] Araujo PR, Yoon K, Ko D, Smith AD, Qiao M, Suresh U, Burns SC, Penalva LOF. Before it gets started: regulating translation at the 5′UTR., 2012, 2012: 475731.
[89] Mayr C. What are 3′UTRs doing?, 2019, 11(10): a034728.
[90] Zhao WX, Pollack JL, Blagev DP, Zaitlen N, McManus MT, Erle DJ. Massively parallel functional annotation of 3′ untranslated regions., 2014, 32(4): 387– 391.
[91] Jia LF, Mao YH, Ji QQ, Dersh D, Yewdell JW, Qian SB. Decoding mRNA translatability and stability from the 5′UTR., 2020, 27(9): 814–821.
[92] Siegel DA, Le Tonqueze O, Biton A, Zaitlen N, Erle DJ. Massively parallel analysis of human 3′UTRs reveals that AU-rich element length and registration predict mRNA destabilization., 2022, 12(1): jkab404.
[93] Pratt BM, Won H. Advances in profiling chromatin architecture shed light on the regulatory dynamics underlying brain disorders., 2022, 121: 153–160.
[94] Dixit A, Parnas O, Li BY, Chen J, Fulco CP, Jerby-Arnon L, Marjanovic ND, Dionne D, Burks T, Raychowdhury R, Adamson B, Norman TM, Lander ES, Weissman JS, Friedman N, Regev A. Perturb-Seq: dissecting molecular circuits with scalable single-cell RNA profiling of pooled genetic screens., 2016, 167(7): 1853–1866.e17.
[95] Klein JC, Agarwal V, Inoue F, Keith A, Martin B, Kircher M, Ahituv N, Shendure J. A systematic evaluation of the design and context dependencies of massively parallel reporter assays., 2020, 17(11): 1083–1091.
[96] Johnson GD, Barrera A, McDowell IC, D’Ippolito AM, Majoros WH, Vockley CM, Wang XY, Allen AS, Reddy TE. Human genome-wide measurement of drug-responsive regulatory activity., 2018, 9(1): 5317.
[97] Penaud-Budloo M, Le Guiner C, Nowrouzi A, Toromanoff A, Chérel Y, Chenuaud P, Schmidt M, von Kalle C, Rolling F, Moullier P, Snyder RO. Adeno-associated virus vector genomes persist as episomal chromatin in primate muscle., 2008, 82(16): 7875–7885.
[98] Duan D, Sharma P, Yang J, Yue Y, Dudus L, Zhang Y, Fisher KJ, Engelhardt JF. Circular intermediates of recombinant adeno-associated virus have defined structural characteristics responsible for long-term episomal persistence in muscle tissue., 1998, 72(11): 8568–8577.
[99] Kvon EZ, Zhu YW, Kelman G, Novak CS, Plajzer-Frick I, Kato M, Garvin TH, Pham Q, Harrington AN, Hunter RD, Godoy J, Meky EM, Akiyama JA, Afzal V, Tran S, Escande F, Gilbert-Dussardier B, Jean-Marçais N, Hudaiberdiev S, Ovcharenko I, Dobbs MB, Gurnett CA, Manouvrier-Hanu S, Petit F, Visel A, Dickel DE, Pennacchio LA. Comprehensive in vivo interrogation reveals phenotypic impact of human enhancer variants., 2020, 180(6): 1262–1271.e15.
[100] Hanson-Smith V, Kolaczkowski B, Thornton JW. Robustness of ancestral sequence reconstruction to phylogenetic uncertainty., 2010, 27(9): 1988–1999.
Massively parallel reporter assay: a novel technique for analyzing the regulation of gene expression
Meng Yuan1,2,3, Hui Li1,2,3, Shouzhi Wang1,2,3
Massively parallel reporter assay (MPRA) is a high-throughput analysis method that can simultaneously investigate the activity of thousands of regulatory elements in the genome. MPRA introduces a uniquely identified barcode on a conventional luciferase reporter gene vector, sequences the DNA barcode before transfection and the mRNA barcode after transfection by next-generation sequencing technology, and uses the ratio of mRNA and DNA barcode reads to analyze the activity of cis-regulatory elements. Since MPRA was proposed, it has been widely used in the identification of genomic cis-regulatory elements and functional variants, the effect of post-transcriptional regulation on phenotypes and so on. In this review, we summarize the development history, basic principles, experimental procedures and statistical analysis methods of MPRA, and its applications in post-transcriptional regulation and cis-regulatory elements. It also provides prospects for its development and useful references for researchers in related fields to understand and apply MPRA.
massively parallel reporter assay; gene expression regulation; cis-regulatory elements; post-transcriptional regulation
2023-07-03;
2023-09-30;
2023-10-07
十四五国家重点研发计划(编号:2022YFF1000201),国家自然科学基金项目(编号:31572394),国家现代农业产业技术体系项目(编号:CARS-41)和黑龙江省自然科学基金联合引导项目(编号:LH2021C036)资助[Supported by the National Key Research and Development Program of China (No. 2022YFF1000201), the National Natural Science Foundation of China (No. 31572394), China Agriculture Research System of MOF and MARA (No. CARS-41), and the Joint Guidance Project of Heilongjiang Natural Science Foundation (No. LH2021C036)]
袁萌,硕士研究生,专业方向:动物遗传育种与繁殖。E-mail: yuanmeng1501@163.com
王守志,博士,教授,博士生导师,研究方向:动物遗传育种与繁殖。E-mail: shouzhiwang@neau.edu.cn
10.16288/j.yczz.23-180
(责任编委: 方向东)
