叶黄素纳米混悬剂制备及其体内药动学研究
2023-10-30陈永顺蒋建平梁建梅贾晓栋
陈永顺,李 静,蒋建平,梁建梅,杨 斌,贾晓栋
(1.商丘医学高等专科学校,河南 商丘 476000; 2.广西医科大学,广西 南宁 530021)
叶黄素是一种含有2 个紫罗酮环的天然类胡萝卜素,广泛存在于胡萝卜、万寿菊等植物中,具有抗氧化、预防心脑血管疾病、保护脑功能、预防白内障等活性[1-2],通过免疫调节、抑制肿瘤细胞生长等机制对皮肤癌、食管癌、前列腺癌、胃癌等有较强活性[2-3],长期毒性研究显示,该成分基本无不良反应,安全性较高[4]。但叶黄素溶解度仅为(6.69±0.10) μg/mL,溶出度较低; 表观油水分配系数为1.03[5],具有一定脂溶性; 相对生物利用度只有2% ~9.4%[6],目前已有脂质体[7]、固体分散体[3]、纳米晶体[8]等相关报道。
前期报道,纳米混悬剂通过降低药物粒径来实现增加溶解度、促进溶出及体内吸收等作用[9-12],黄雅兰等[9]以十六烷基三甲基氯化钠为乳化剂制备了叶黄素纳米晶体,但它有一定毒性[13],需加入大量冻干保护剂制成冻干粉,从而增加了辅料种类及用量。白蛋白是一种天然乳化剂,安全性高,本身还可作为冻干保护剂[14],可减少辅料用量,故本实验以牛血清白蛋白为稳定剂制备叶黄素纳米混悬剂[15-17],并考察其体内药动学,以期为相关新型制剂开发提供新的思路。
1 材料
Agilent 1100 型高效液相色谱仪(美国Agilent公司); CD-X15 型超声仪(深圳市亦为实业有限公司); JB-1 型磁力搅拌器(上海叶拓科技有限公司); MSE125P-CE 型电子天平(德国Sartorius 公司); YSB-BCF12 型超低温冰箱(佛山市雅绅宝制冷设备制造有限公司); RC-6D 型溶出仪(天津创兴电子设备制造股份有限公司); BMH 型真空冻干机(北京博劢设备有限公司); Master-sizer 型粒度分析仪(英国马尔文仪器有限公司); HAC-I 型氮吹仪(北京莱宁德锐科技有限公司)。
叶黄素对照品(批号200510,纯度92%,上海如吉生物科技有限公司); 叶黄素原料药(批号20200812,纯度90%,成都彼样生物科技有限公司); 麦角甾醇对照品 (批号20220522,纯度98%,成都嘉叶生物科技有限公司)。吐温-80 (批号20201212,天津红太阳化工有限公司); 牛血清白蛋白(BSA,批号20201118,上海蓝季生物科技有限公司); 十二烷基硫酸钠(SDS,批号191006,国药集团化学试剂有限公司); 甘露醇 (批号190915,湖南九典制药股份有限公司)。
SD 大鼠,雌雄兼具,体质量 (220±20) g,购自河南省实验动物中心,动物生产许可证号SCXK (豫) 2020-0012。
2 方法与结果
2.1 叶黄素含量测定
2.1.1 色谱条件 Hypersil ODS C18色谱柱(250 mm×4.6 mm,5 μm); 流动相甲醇-乙腈-二氯甲烷(55 ∶ 25 ∶ 20); 体积流量1.0 mL/min; 柱温35 ℃; 检测波长446 nm; 进样量10 μL。
2.1.2 供试品溶液制备 取纳米混悬剂1 mL,置于50 mL 量瓶中,加入40 mL 甲醇超声处理5 min,甲醇稀释至刻度,0.45 μm 微孔滤膜过滤,精密量取2 mL 至10 mL 量瓶中,流动相稀释至刻度,即得。
2.1.3 对照品溶液制备及线性关系考察 取叶黄素对照品适量,甲醇制成质量浓度为200 μg/mL的贮备液,流动相依次稀释至5、2.5、1、0.5、0.1、0.05 μg/mL,即得。以对照品质量浓度为横坐标(X),峰面积为纵坐标(Y) 进行回归,得方程为Y=715.84X+126.5 (r=0.999 8),在0.05 ~5 μg/mL 范围内线性关系良好。
2.1.4 方法学考察 取0.05、1、5 μg/mL 对照品溶液适量,在“2.1.1” 项色谱条件下进样测定6次,测得叶黄素含量RSD 分别为0.22%、0.60%、0.43%,表明仪器精密度良好。取纳米混悬剂适量,按“2.1.2” 项下方法平行制备6 份供试品溶液,在“2.1.1” 项色谱条件下进样测定,测得叶黄素含量RSD 为1.70%,表明该方法重复性良好。取供试品溶液适量,室温下于0、3、6、9、12、24 h 在“2.1.1” 项色谱条件下进样测定,测得叶黄素含量RSD 为0.48%,表明溶液在24 h 内稳定性良好。取9 份纳米混悬剂,每份0.5 mL,置于50 mL 量瓶中,分为低、中、高3 组,分别加入对照品贮备液1、2、3 mL,按“2.1.2” 项下方法制备供试品溶液,在“2.1.1” 项色谱条件下进样测定,测得叶黄素平均加样回收率分别为100.63%、99.47%、101.08%,RSD 分别为0.36%、0.89%、0.77%。
2.2 纳米混悬剂制备 参考文献[17-18] 报道,取叶黄素原料药30 mg,溶于5 mL 四氢呋喃中,作为有机相; 配制一定浓度含吐温-80、牛血清白蛋白的水溶液50 mL,45 ℃水浴加热,作为水相,将有机相缓慢滴入水相中,800 r/min 磁力搅拌40 min,置于均质机中循环均质,0.45 μm 微孔滤膜过滤,蒸馏水补足至50 mL,置于冰水混合物中降温,即得。
2.3 粒径、PDI、Zeta 电位测定 取纳米混悬剂0.5 mL,蒸馏水稀释50 倍,涡旋10 s 混匀,在粒度分析仪上测定,平行3 次,取平均值。
2.4 处方优化 采用单因素试验。
2.4.1 白蛋白浓度 固定叶黄素用量30 mg,吐温-80 浓度0.5%,均质压力80 MPa,均质次数6次,考察白蛋白浓度对粒径、PDI 的影响,结果见图1。由此可知,处方中仅有吐温-80 时粒径接近1 000 nm,PDI 大于0.6; 随着白蛋白浓度增加粒径先降低后趋稳,而PDI 先降低后升高,为1.5%时两者最低。最终确定,白蛋白浓度为1.5%。
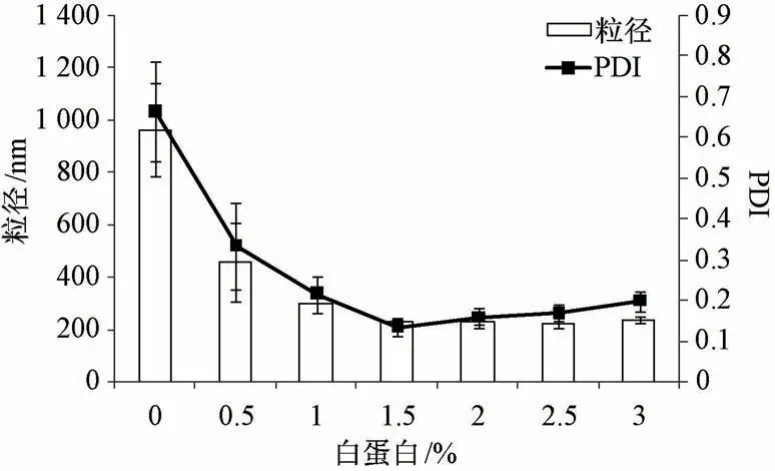
图1 白蛋白浓度对粒径、PDI 的影响(n=3)Fig.1 Effects of albumin concentration on particle size and PDI (n=3)
2.4.2 吐温-80 浓度 固定叶黄素用量30 mg,白蛋白浓度1.5%,均质压力80 MPa,均质次数6次,考察吐温-80 浓度对粒径、PDI 的影响,结果见图2。由此可知,白蛋白浓度为1.5% (不加吐温-80) 时粒径约为1 500 nm,PDI 大于0.7; 随着吐温-80 浓度增加粒径、PDI 均先降低后趋稳,大于0.75%时两者变化不明显。最终确定,吐温-80浓度为0.75%。
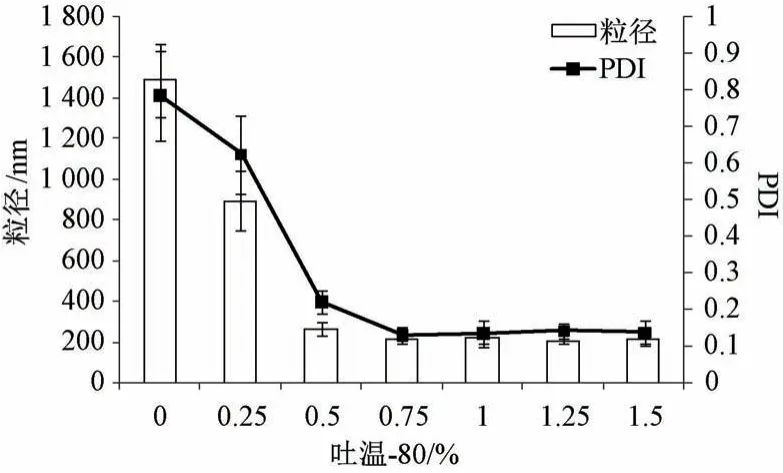
图2 吐温-80 浓度对粒径、PDI 的影响(n=3)Fig.2 Effects of Tween-80 concentration on particle size and PDI (n=3)
2.4.3 均质压力 固定叶黄素用量30 mg,白蛋白浓度1.5%,吐温-80 浓度0.75%,均质次数6次,考察均质压力对粒径、PDI 的影响,结果见图3。由此可知,随着均质压力增加粒径、PDI 均先降低后升高,可能是因为压力较小时无法提供足够能量形成较小粒径的纳米粒,而过大时可能会导致体系温度较高,破坏纳米粒保护层[18-19]并发生融合、聚集,导致两者变大。最终确定,均质压力为80 MPa。
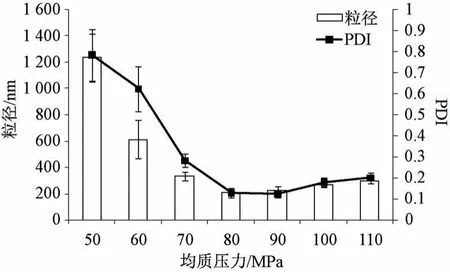
图3 均质压力对粒径、PDI 的影响(n=3)Fig.3 Effects of homogenization pressure on particle size and PDI (n=3)
2.4.4 均质次数 固定叶黄素用量30 mg,白蛋白浓度1.5%,吐温-80 浓度0.75%,均质压力80 MPa,考察均质次数对粒径、PDI 的影响,结果见图4。由此可知,随着均质次数增加粒径、PDI均先降低后升高,可能是因为次数过多时体系温度急剧升高,纳米粒之间容易发生融合、聚集,粒径分布不均匀,导致两者变大,并且为8 次时均降低。最终确定,均质次数为8 次。
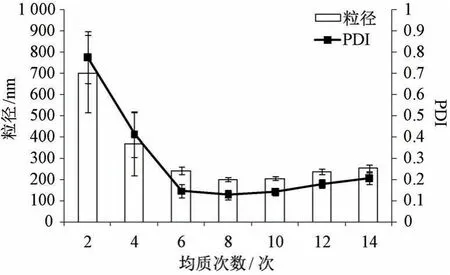
图4 均质次数对粒径、PDI 的影响(n=3)Fig.4 Effects of homogenization frequency on particle size and PDI (n=3)
2.5 验证试验 “2.4” 项下结果显示,最优处方为叶黄素用量30 mg,白蛋白浓度1.5%,吐温-80 浓度0.75%,均质压力80 MPa,均质次数8 次,平均粒径为(208.71±9.26) nm,PDI 为0.114±0.017,Zeta 电位为- (23.15±1.60) mV,见图5~6。
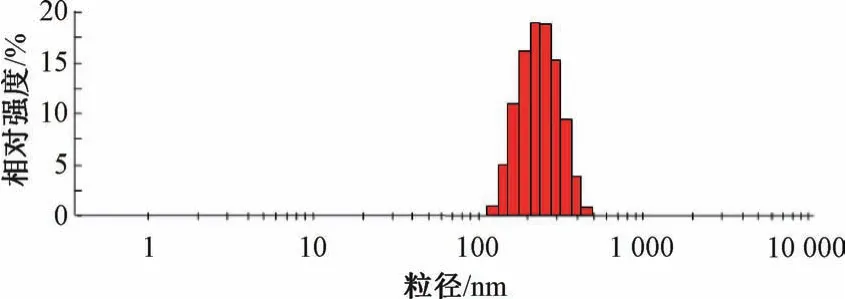
图5 叶黄素纳米混悬剂粒径分布Fig.5 Particle size distribution of lutein nanosuspensions
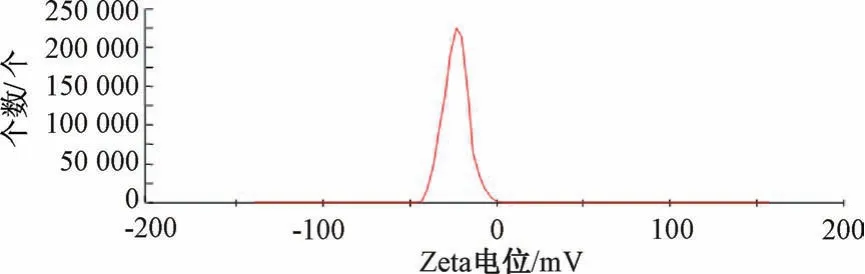
图6 叶黄素纳米混悬剂Zeta 电位Fig.6 Zeta potential of lutein nanosuspensions
2.6 形态观察 取纳米混悬剂0.5 mL,蒸馏水稀释50 倍,混匀,滴于铜胶带上,室温晾干,喷金1 min,再按处方工艺制备空白样品(不含叶黄素,其余辅料比例同纳米混悬剂),同法测定,在扫描电镜(放大15 000 倍) 下观察,结果见图7。由此可知,纳米混悬剂基本呈球形,而空白样品(不含叶黄素) 中未发现纳米粒。

图7 叶黄素纳米混悬剂(A)、空白样品(B) 扫描电镜图Fig.7 Scanning electron microscopy images of lutein nanosuspensions (A) and blank sample (B)
2.7 冻干粉制备及表征
2.7.1 制备工艺 取纳米混悬剂适量,加入一定质量分数甘露醇,涡旋30 s 至澄清,分装至西林瓶中(每份均3 mL),密封置于-30 ℃冰箱中预冻2 d后立即转移至冷冻干燥机(冷阱温度-55 ℃)中,当真空度达到0.01 kPa 时冻干2 d,取出,即得,蒸馏水复溶,考察甘露醇用量对粒径、PDI 的影响,结果见图8。由此可知,不添加甘露醇时冻干粉复溶后粒径为(252.67±18.13) nm,PDI 为(0.132±0.013),可能与白蛋白本身是一种优良冻干保护剂有关,但其外观很差; 随着甘露醇用量增加冻干粉外观逐渐达饱满状态,但超过2%时粒径有增加趋势,超过3%时PDI 也开始变大。最终确定,以1.5%甘露醇为冻干保护剂,此时冻干粉外观饱满,复溶后粒径、PDI 较小。
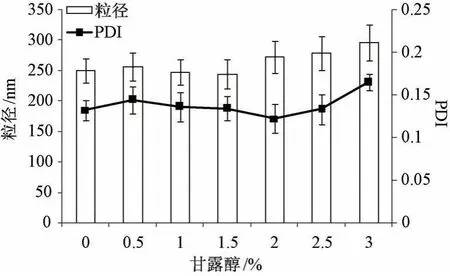
图8 甘露醇用量对粒径、PDI 的影响(n=3)Fig.8 Effects of mannitol consumption on particle size and PDI (n=3)
2.7.2 晶型分析 设定条件为扫描范围3°~45°,电压40 kV,扫描速度6°/min,Cu-Kα 靶。取原料药、空白辅料(不含药物,辅料比例同纳米混悬剂冻干粉)、物理混合物、新鲜纳米混悬剂冻干粉、放置6 个月纳米混悬剂冻干粉(温度30 ℃,相对湿度65%) 适量,进行X 射线粉末衍射(XRPD) 分析,结果见图9。由此可知,原料药在20.3°、28.4°、40.2°处出现特征晶型峰[4]; 由于辅料掩蔽作用,物理混合物图谱中仅发现原料药在40.2°处的晶型峰,表明它以晶型状态存在; 新鲜纳米混悬剂冻干粉图谱中未发现原料药晶型峰,表明它可能转变为无定形状态; 放置6 个月纳米混悬剂冻干粉图谱中仍发现原料药晶型峰,表明其稳定性良好。
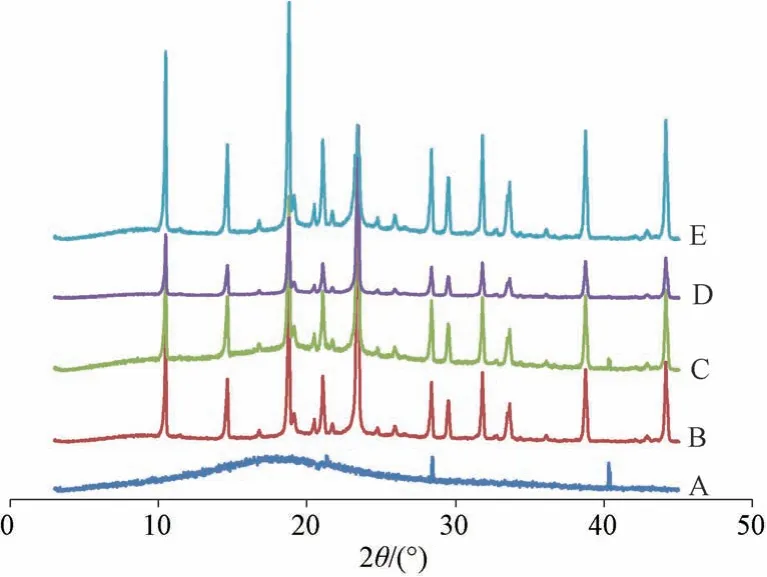
图9 各样品XRPD 图Fig.9 XRPD patterns for various samples
2.8 溶解度测定 取过量原料药、物理混合物、纳米混悬剂冻干粉至烧杯中,加入10 mL 蒸馏水,在25 ℃水浴中800 r/min 磁力搅拌72 h,取混悬液,0.45 μm 微孔滤膜过滤,取续滤液,测定溶解度。结果,原料药、物理混合物溶解度分别为(6.69±0.10)、(6.94±0.11) μg/mL,而纳米混悬剂冻干粉达(308.57±2.02) μg/mL,即与原料药相比增加至46.12 倍。
2.9 体外溶出研究 由于叶黄素在1.5%SDS 溶液中的溶解度为46.47 μg/mL,故10 mg 该成分在1 000 mL 1.5%SDS 溶液中即可达漏槽条件。取纳米混悬剂冻干粉(含10 mg 叶黄素) 适量,加入1.5%SDS 溶液5 mL,置于透析袋中(截留分子量8 000 ~12 000 Da),扎紧,设定释药介质为1 000 mL 1.5% SDS 溶液(温度37 ℃),转速为75 r/min,于0、5、15、30、45、60、90、120、240、360、480 min 各取样5 mL,并补加5 mL 空白介质,0.45 μm 微孔滤膜过滤,取续滤液,测定溶出度,同法测定原料药、物理混合物,结果见图10。由此可知,原料药在漏槽条件下480 min 内溶出度仅为19.80%,可能与其粒度较大、疏水性较强有关[19],而纳米混悬剂冻干粉在360 min 内累积溶出度即达95%。
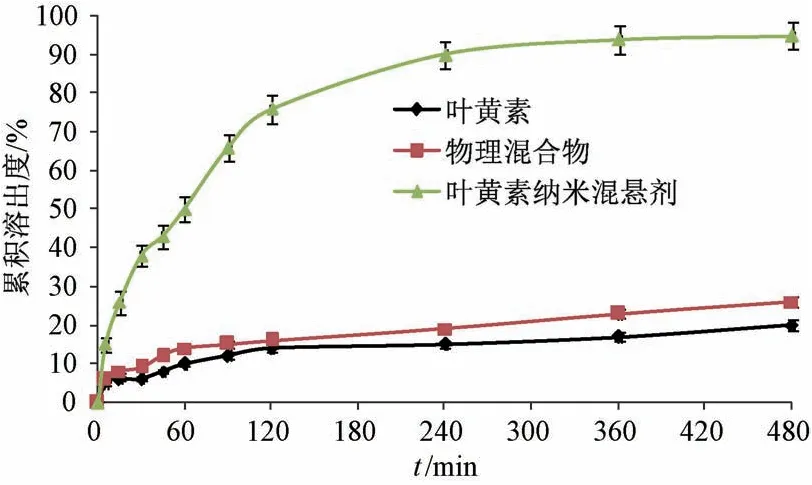
图10 各样品体外溶出曲线(n=6)Fig.10 In vitro dissolution curves for various samples (n=6)
2.10 稳定性研究 取纳米混悬剂冻干粉适量,蒸馏水复溶,测定离心前粒径,再取2 mL 至离心管中,1 500 r/min 离心30 min,取上层混悬液测定离心后粒径,计算稳定系数[20],公式为稳定系数=离心后粒径/离心前粒径(越接近1,样品越稳定),然后置于温度30 ℃、相对湿度65% 的恒温恒湿箱中,于0、3、6、9、15、30、45、60、90 d 同法测定,结果见图11。由此可知,纳米混悬剂冻干粉放置90 d 后稳定系数仍大于0.95,表明其稳定性良好。
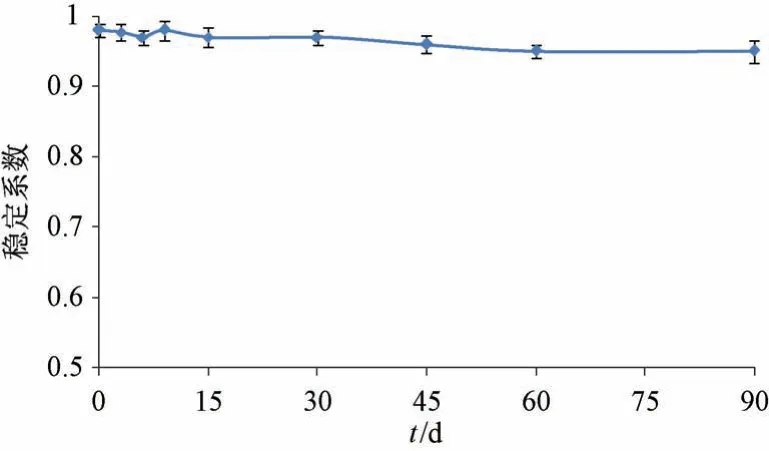
图11 叶黄素纳米混悬剂不同时间点稳定系数(n=3)Fig.11 Stability coefficients of lutein nanosuspensions at different time points (n=3)
2.11 体内药动学研究
2.11.1 分组、给药与采血 取原料药、物理混合物、纳米混悬剂冻干粉适量,0.5% CMC-Na 溶液制成药液(以叶黄素计,质量浓度为4 mg/mL)。18 只大鼠随机分为3 组,每组6 只,按30 mg/kg剂量灌胃给药,于0.5、1、1.5、2、2.5、3、4、6、12、18 h 眼眶静脉丛取血各0.25 mL,置于肝素浸润离心管中,密封,涡旋5 s 混匀,3 000 r/min离心2 min,取出血浆,冷冻保存。
2.11.2 血浆样品处理 参考文献[21] 报道,取麦角甾醇对照品适量,乙腈稀释至886 ng/mL,作为内标溶液,取50 μL,加到100 μL 血浆样品中,涡旋混匀1 min,加入1 mL 氯仿,涡旋3 min,4 ℃、10 000 r/min 离心10 min,收集氯仿层,40 ℃氮吹仪吹干,100 μL 乙腈复溶,10 000 r/min离心10 min。
2.11.3 线性关系考察 取叶黄素对照品适量,乙腈制成2 080、1 040、520、260、104、52 ng/mL溶液,分别取100 μL 至离心管中,40 ℃氮吹仪吹干,残渣中加入100 μL 空白血浆,涡旋3 min,按“2.11.2” 项下方法处理,即得质量浓度分别为2 080、1 040、520、260、104、52 ng/mL 的血浆对照品溶液(含内标)。以对照品质量浓度为横坐标(X),叶黄素、麦角甾醇峰面积比值为纵坐标(Y) 进行回归,得方程为Y=0.001 9X-0.236 7(r=0.995 4),在52 ~2 080 ng/mL 范围内线性关系良好。
2.11.4 方法学考察 取52、520、2 080 ng/mL血浆对照品溶液适量,同一天内在“2.1.1” 项色谱条件下进样测定6 次,测得叶黄素、麦角甾醇峰面积比值RSD 分别为6.05%、5.26%、5.06%,表明该方法日内精密度良好; 同法连续测定6 d,每天1 次,测得两者峰面积比值RSD 分别为9.43%、6.00%、7.52%,表明该方法日间精密度良好。取给药0.5 h 后血浆样品适量,于0、3、6、9、12、24 h 在“2.1.1” 项色谱条件下进样测定,测得叶黄素、麦角甾醇峰面积比值RSD 为7.44%,表明样品在24 h 内稳定性良好。取上述3 种质量浓度血浆对照品溶液适量,按“2.11.2” 项下方法处理,在“2.1.1” 项色谱条件下进样测定,测得叶黄素平均加样回收率分别为93.67%、96.08%、93.82%,RSD 分别为6.20%、7.35%、4.69%。
2.11.5 结果分析 血药浓度-时间曲线见图12,主要药动学参数见表1。由此可知,原料药、物理混合物相关数值比较,差异无统计学意义 (P>0.05),表明辅料对原料药口服吸收基本无影响;与原料药、物理混合物比较,纳米混悬剂tmax缩短(P<0.05),Cmax、AUC0~t、AUC0~∞升高 (P<0.01),相对生物利用度相较于原料药增加至3.71倍,但t1/2无明显变化(P>0.05)。
表1 叶黄素主要药动学参数(±s,n=6)Tab.1 Main pharmacokinetic parameters for lutein (±s,n=6)
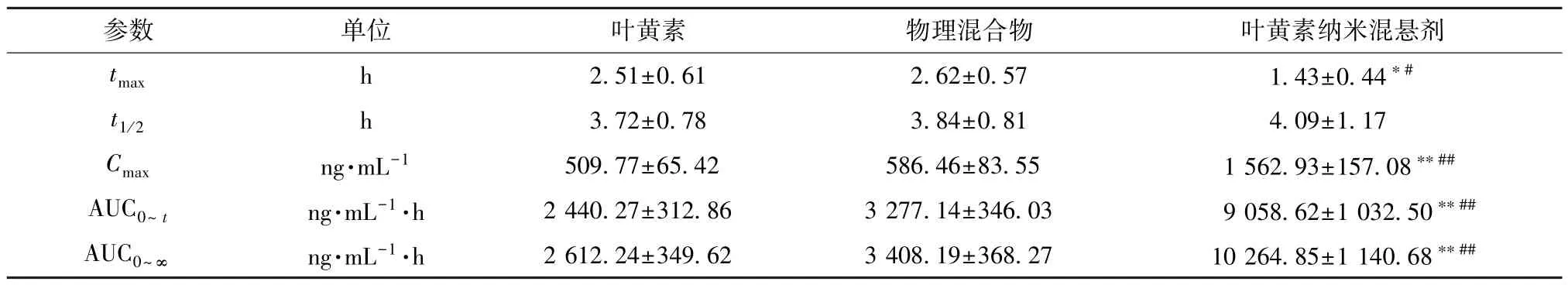
表1 叶黄素主要药动学参数(±s,n=6)Tab.1 Main pharmacokinetic parameters for lutein (±s,n=6)
注: 与叶黄素比较,*P<0.05,**P<0.01; 与物理混合物比较,#P<0.05,##P<0.01。
参数单位叶黄素物理混合物叶黄素纳米混悬剂tmaxh2.51±0.612.62±0.571.43±0.44*#t1/2h3.72±0.783.84±0.814.09±1.17 Cmaxng·mL-1509.77±65.42586.46±83.551 562.93±157.08**##AUC0~tng·mL-1·h2 440.27±312.863 277.14±346.039 058.62±1 032.50**##AUC0~∞ng·mL-1·h2 612.24±349.623 408.19±368.2710 264.85±1 140.68**##
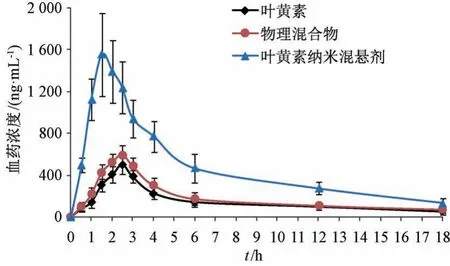
图12 叶黄素血药浓度-时间曲线(n=6)Fig.12 Plasma concentration-time curves for lutein (n=6)
3 讨论与结论
白蛋白吸附在纳米粒表面并发生交联后形成界面膜,伸向水相的长链提供了巨大的空间位阻,从而起到稳定纳米粒的作用[22]。为了减小纳米混悬剂粒径,本实验在处方中引入非离子表面活性剂吐温-80[23]。另外,处方中白蛋白除了作为稳定剂外,本身也是一种冻干保护剂,只需1.5%甘露醇即可制得外观饱满的冻干粉,大大降低了辅料用量。
白蛋白在均质条件下可形成纳米粒[12-13,24],但本实验按处方工艺制备的叶黄素纳米混悬剂空白样品中并未发现纳米粒,可能是由于吐温-80 通过氢键结合到白蛋白疏水区,增强了后者稳定性,从而阻止其变性形成纳米粒[25-26]。另外,叶黄素纳米混悬剂中原料药晶型发生了变化,可能是除去有机溶剂时后者逐渐析出,此时稳定剂的空间稳定作用、缠绕作用、静电作用等抑制了结晶形成,最终使其以无定形状态存在[9]。
体内药动学研究结果显示,叶黄素纳米混悬剂tmax显著缩短,可能与其前期释药速率较快有关;Cmax、相对生物利用度分别增加至3.06、3.71 倍,可能是由于纳米混悬剂可大大提高原料药溶解度、累积溶出度,解决了吸收瓶颈: 纳米混悬剂可使原料药比表面积更大,增加与胃肠道的接触面,有利于吸收[23]。另外,叶黄素属于生物药剂学Ⅳ药物[5],尽管纳米混悬剂有效改善了其水溶性、溶出度,但可能对脂溶性的影响有限,故会限制其生物利用度的提高程度。
综上所述,本实验完成了叶黄素纳米混悬剂的制备及其体内药动学的考察,可为后续该制剂相关剂型(片剂或胶囊剂) 开发、药效学评价等方面奠定坚实基础。
