新疆冬小麦品种资源主要产量性状全基因组关联分析
2023-10-23马艳明娄鸿耀张胜军王威郭营倪中福刘杰
马艳明,娄鸿耀,张胜军,王威,郭营,倪中福,刘杰
新疆冬小麦品种资源主要产量性状全基因组关联分析
马艳明,娄鸿耀3,张胜军4,王威1,郭营5,倪中福2,刘杰
1新疆农业科学院农作物品种资源研究所,乌鲁木齐 830091;2中国农业大学农学院,北京 100193;3北京市农林科学院杂交小麦研究所,北京 100097;4新疆伊犁哈萨克自治州农业科学研究所,新疆伊宁 835011;5山东农业大学农学院,山东泰安 271018
【目的】高产是小麦育种的永恒主题,利用全基因组关联分析发掘控制小麦产量性状的QTL区段及优异基因,为小麦分子标记辅助选择育种提供理论依据和标记信息。【方法】以新疆本地188个冬小麦品种资源为材料,利用小麦55K SNP芯片进行全基因组扫描,通过对6个不同环境下的株高、穗长、小穗数、可育小穗数、穗粒数、千粒重、粒长、粒宽、籽粒长/宽比9个产量相关性状进行表型鉴定,利用6个环境下各性状数据及最佳线性无偏预测(BLUP)数据,基于混合线性模型(MLM)对表型和基因型进行全基因组关联分析。【结果】经主成分分析,将188个材料分为地方品种和育成品种2个亚群;利用6个环境下各性状数据,9个性状共检测到1 309个显著性SNP标记,其中,每个显著性SNP位点可解释7.259%—70.792%的表型变异。利用BLUP数据,9个性状共检测到66个显著性位点,同时与2个性状关联的共有SNP位点有5个,贡献率波动范围为8.498%—21.877%。将同时与2个性状或2个以上环境关联到的重复位点作为稳定的显著性关联位点,9个性状共检测到38个稳定关联位点,包括株高重复位点5个,穗长重复位点10个,小穗数重复位点10个,结实小穗数重复位点6个,穗粒数重复位点6个,千粒重重复位点1个,可解释9.10%—23.81%的表型变异。将这38个位点与已发布小麦基因组位点比对,仅找到3个与本研究关联重复位点位置相近、且有注释基因功能的基因,分别是:2A染色体上与株高关联位点AX-108794050距离相近的,与转录因子bHLH71的代谢合成有关;1A染色体上与穗长关联位点AX-110689765距离相近的,与蛋白质编码有关;4B染色体上与千粒重关联位点AX-110399975距离相近的,与编码丝氨酸/苏氨酸蛋白激酶SD1-8有关,参与对细胞增殖与分化的调控。【结论】检测到38个与小麦产量性状关联的QTL位点,关联的优良等位基因具有降低株高、增加穗长、小穗数、可育小穗数、穗粒数和千粒重的作用。
小麦;产量性状;SNP标记;关联分析;候选基因
0 引言
【研究意义】普通小麦(L.)是典型的异源多倍体口粮作物。中国是全球最大的小麦生产国和消费国,其产量常年占全球总产量的17%。因此,我国小麦产量的持续提升对保障国家粮食安全具有非常重要的意义,同时也在一定程度上影响国际粮价的稳定[1]。随着耕地面积的不断减少和全球总人口数量的不断增加,高产育种一直是小麦育种工作最为重要的内容之一。而株高、穗长、小穗数、可育小穗数、穗粒数、千粒重以及粒长、粒宽、籽粒长宽比等表现都与小麦的产量存在紧密的联系。在传统育种中,育种家主要依靠经验对上述产量相关性状进行表型选择,其选择效率往往较低,且容易受到环境因素的影响,造成选择的误差。利用分子手段,鉴定与产量性状相关联的重要遗传位点,并根据这些位点开发出育种可用的分子标记,可显著提升对小麦产量相关性状选择的效率和准确性,大幅缩短新品种选育的周期[2]。【前人研究进展】近年来,应用关联分析发掘粮食作物产量相关QTL(quantitative trait loci)已成为作物基因组学研究的热点之一。随着测序技术的迅猛发展和统计算法的革新,全基因组关联分析(genome-wide association study,GWAS)已经成为鉴定作物产量相关QTL的重要方法。该方法不仅能够提高双亲分离群体作图的分辨率,同时还能对多个农艺性状同时进行关联作图,提高了QTL鉴定的效率[3]。作为异源多倍体作物,普通小麦基因组复杂度高,给GWAS工作带来一定困难。尽管如此,在过去的研究中,国内外学者已经利用GWAS鉴定出一些与小麦产量性状相关的多态性位点,但在不同的实验条件下,其关联分析的结果还存在不一致的现象[4]。Qaseem等[5]对12种与产量相关的农艺性状进行GWAS分析,共获得114个与性状关联的单核苷酸多态性位点(single nucleotide polymorphism,SNP),其中21个位点与单株穗数相关联,4个位点与单穗粒数相关联,7个位点与千粒重相关联;SUKUMARAN等[6]利用90K SNP芯片对287个小麦材料进行全基因组扫描,检测到与粒重、亩穗数、成熟期关联的位点;Chen等[7]利用SNP标记对205份小麦品种进行全基因组关联分析,得到271个与千粒重等特征相关联的标记。Sun等[8]利用20 689个高质量小麦90K SNP标记,针对163个小麦品种在3个地点14个环境中的13个产量相关性状进行分析,关联得到1 769个重要位点,其中有41个SNP位点可能对产量相关性状的形成有重要贡献。Wang等[9]对105个小麦品种进行产量相关性状的GWAS分析,共找到24个与9种产量相关性状连锁的标记,其中,与千粒重、籽粒形态和植株高度相关的4个位点在不同环境中均被检测到,认为这些位点受环境影响较小,具有重要的育种利用价值;而有效穗数、籽粒总产量、叶面积等性状受环境影响较大;染色体上的同一区域或邻近区域可能与多个产量相关性状关联,表明一因多效性。Ye等[10]利用55K SNP芯片对244个四川小麦品种(包括79个地方品种和165个审定品种)的产量性状进行分析,关联得到6个穗长相关QTL和3个千粒重相关QTL,通过与已知QTL的物理位置进行比较,鉴定出21个与小麦生长代谢相关的候选基因。翟俊鹏等[11]利用小麦35K SNP芯片对150份小麦品种在4个环境条件下的9个主要农艺性状进行全基因组关联分析,检测得到652个关联位点,其中21个位点在2个以上(包括2个)的环境中被重复检测到。赵丹阳等[12]结合SSR与SNP标记对52个黄淮麦区品种(系)材料的株高、穗长、单株穗数、可育小穗数、穗粒数、千粒重等6个产量性状进行标记分析,获得6个与产量性状显著关联的SSR标记和7个与产量性状相关的SNP位点。顾晶晶等[13]以660K SNP芯片对198份河南小麦品种在4个环境下的穗粒数、穗长、总小穗数、可育小穗数和不育小穗数进行全基因组关联分析,得到41个显著关联位点,并对关联位点进一步分析发掘出多个与穗粒数性状相关的优异等位变异,其单个关联位点的表型变异贡献率范围为6.19%—20.83%。【本研究切入点】小麦产量性状是复杂的多基因控制的数量性状,目前,已知基因标记资源有限,急需挖掘更多与产量性状相关的优异基因。前期利用小麦55K SNP芯片对新疆本地收集的134份冬小麦地方品种和54份育成品种进行了基因型分析,获得了丰富的SNP信息[14]。【拟解决的关键问题】本研究进一步对这些小麦材料在2年3点共计6个环境条件下的株高、穗长、小穗数、可育小穗数、穗粒数、千粒重、粒长、粒宽、籽粒长/宽等9个产量相关性状进行系统表型分析,并结合芯片数据开展全基因组关联分析,旨在发掘与小麦产量相关性状密切相关的QTL位点和基因组区段,为小麦分子标记辅助选择育种提供理论依据和标记信息。
1 材料与方法
1.1 试验材料及表型鉴定
供试材料为188份新疆冬小麦品种资源,包括地方品种134个,育成品种54个[14],均为国家作物种质资源库(新疆分库)收集保存。
供试材料分别于2017年、2018年2个小麦生育期种植于3个试验点,分别是新疆农业科学院安宁渠综合试验场(新疆乌鲁木齐市,UR;44.31°N,86.22°E)、新疆伊犁哈萨克自治州农业科学研究所(新疆伊宁市,YN;43.92°N,81.32°E)、山东农业大学农学实验站(山东泰安市,TA;36.17°N,117.17°E),共计6个环境,分别标记为UR2017、UR2018、YN2017、YN2018、TA2017、TA2018。试验采取完全随机区组设计,每小区种植50个材料,每材料2行,行长1.5 m,每行点播30粒,3次重复,参照《小麦种质资源描述规范和数据标准》[15]规定的方法调查株高、穗长、小穗数、结实小穗数、穗粒数等性状,利用万深SC-G型自动种子考种分析仪测定千粒重、粒长、粒宽和籽粒长/宽,每材料重复测定2次。所有性状均取平均值作为最终测量值。
1.2 基因组DNA提取与SNP标记分析
全部试验材料采用2015年10月种植、2016年7月收获的种子。于2016年10月在中国农业大学小麦遗传育种温室种植,植株长至二叶一心时,取小麦叶片,按Saghai-Maroof等[16]的CTAB法提取基因组DNA。DNA提取后质控其纯度和浓度,并将其稀释至50 ng·μL-1备用。将制备好的188份新疆冬小麦品种基因组DNA送至博奥晶典公司进行55K SNP全基因芯片扫描,获得原始芯片测序数据,质控后获得50 743个SNP多态性位点。
1.3 数据分析
群体结构通过STRUCTURE 2.3.4软件[17]的MCMC模型(the Bayesian Markov Chain Monte Carlo model)估算,设定群体数量为1—10,设置burn-in步长为10 000,MCMC迭代次数为100 000,对于每一个K值,分别进行5次独立的重复,根据数据对数概率(LnP(D))随连续K值的变化率计算Δ,研究材料的类群个数通过Δ确定[18],多次运行结果用CLUMPP软件[19]整合。在群体结构、亲缘关系和连锁不平衡(linkage disequilibrium,)分析基础上,以群体结构作为固定效应,个体亲缘关系作为随机效应,校正群体结构和个体间亲缘关系的影响,采用Tassel 5.0(http://www.maizegenetics. net/)软件中的混合线性模型(mixed linear model,MLM)进行性状与标记间的关联分析,MLM模型同时把亲缘关系与群体结构作为协变量,具有更高的统计效率且降低假阳性发生率[20],获得可靠的与目标性状显著关联的SNP位点,同时,利用多环境下各性状数据及最佳线性无偏预测(best linear unbiased prediction,BLUP)数据进行关联分析,并对关联结果进行比较。BLUP对多环境数据进行整合,去除环境效应,可以得到个体稳定遗传的表型。为尽可能消除关联分析中带来的假阳性结果,采用Bonferroni法[21]对值进行校正。计算公式如下:
= α/N
其中,α=1,N为所用SNP标记数目[22]。50 743个SNP标记的值为1/50743,相当于阈值-log10()≈5。当SNP位点的<0.00001时,视该SNP位点与性状显著关联。把与性状显著关联的SNP标记定位到小麦的基因组上,确定相对应的置信区域,在关联SNP标记连锁不平衡衰减距离的区域范围内,搜寻与性状相关的候选基因。用Structure 2.3.4软件计算Q值,用SPAGedi软件[23]计算品种间亲缘关系的Kinship (K)值,基于公式=α+Qβ+Kµ+e(为基因型,为表型),计算最终每个SNP位点所对应的关联值,运用R3.4.2绘制manhattan图和quantile-quantile图,使显著性位点可视化。用2值来鉴定两位点间的连锁不平衡程度。当2对应<0.01时,认为两位点处于连锁不平衡状态[24]。
2 结果
2.1 群体结构分析
为了准确估计本研究所用自然群体的群体结构,采用K值分析法和主成分分析法进行群体结构分析,以确定最合适的分群数量。利用Structure2.3.4软件对188个小麦品种的群体结构进行系统解析。设定类群数(K值)为1—10,对于每个指定的K值,LnP(D)值随着K值的增加持续上升,在K=2时出现明显拐点,此时值达到最大值(图1-a和图1-c),表明188个小麦品种可以划分为2个亚群,一个是地方品种群,另一个是育成品种群。基于SNP,利用Tassel 5.0软件进行主成分分析(principal components analysis,PCA),前2个主成分PC1(25.34%)、PC2(9.22%)可解释34.56%的群体信息,同样证明全部材料可以分为地方品种和育成品种2个亚群(图1-b),与所选材料一致。
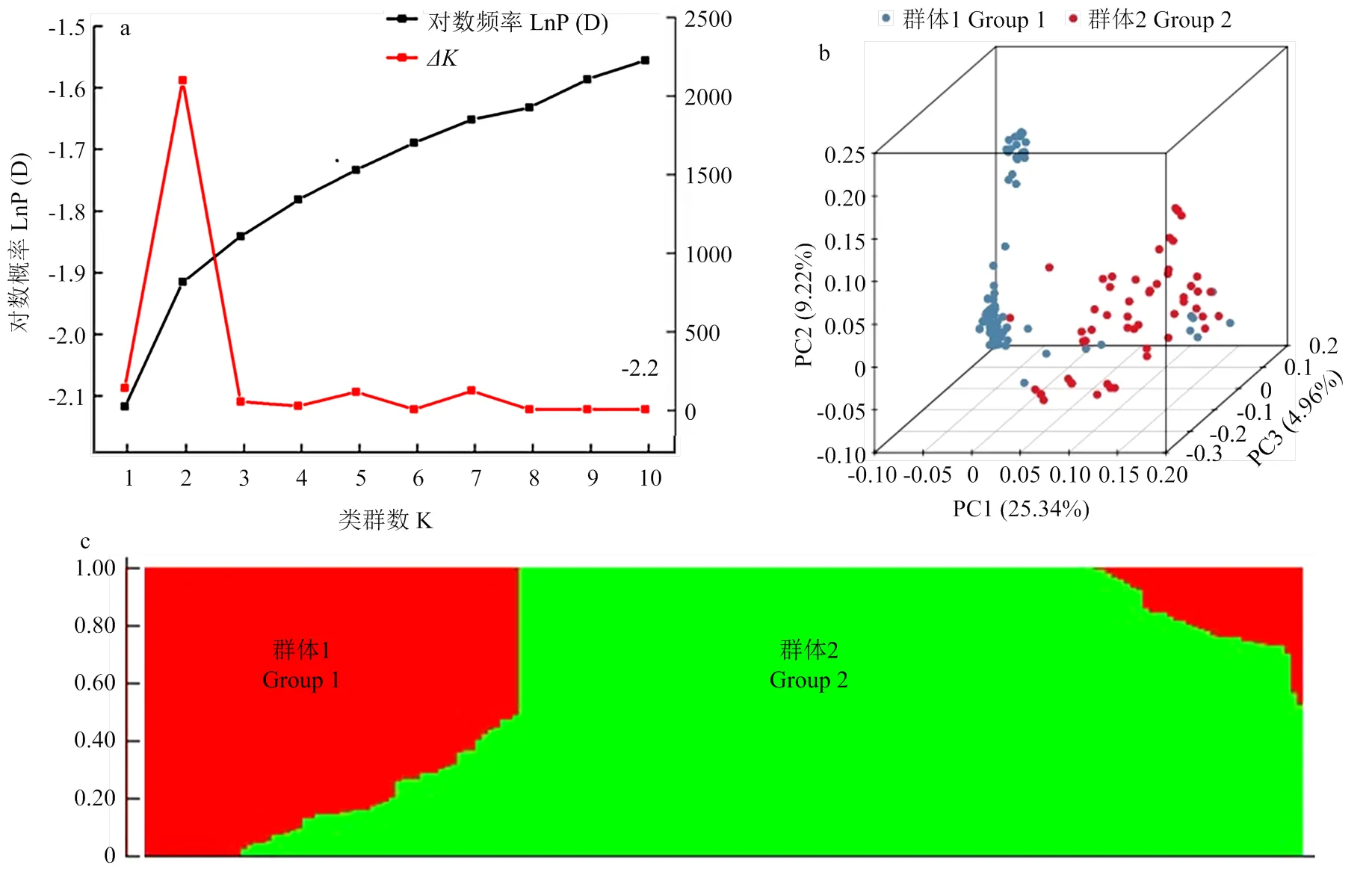
a:K值对应的ΔK值;b:主成分分析;c:基于模型的群体结构分析 a: The ΔK statistic for each given K; b: PCA; c: Model-based ancestries
2.2 品种间的亲缘关系及LD分析
利用SPAGeDi评估品种之间的相对亲缘关系。亲缘关系系数等于0的占54.4%,小于0.1的占31.0%(图2-a),表明群体中大多数种质与其他种质的亲缘关系较弱,群体材料适宜进行关联分析。将过滤后的SNP标记数据用于计算连锁不平衡(LD)。A、B和D亚基因组以及整个基因组的平均2值随着成对标记距离的增加而逐渐降低。当2的临界阈值定义为0.25时,相比之下,A基因组的LD衰减距离最小(约5 Mb),B基因组和D基因组LD衰减距离较大(>15 Mb),整个基因组LD衰减距离约为15 Mb(图2)。
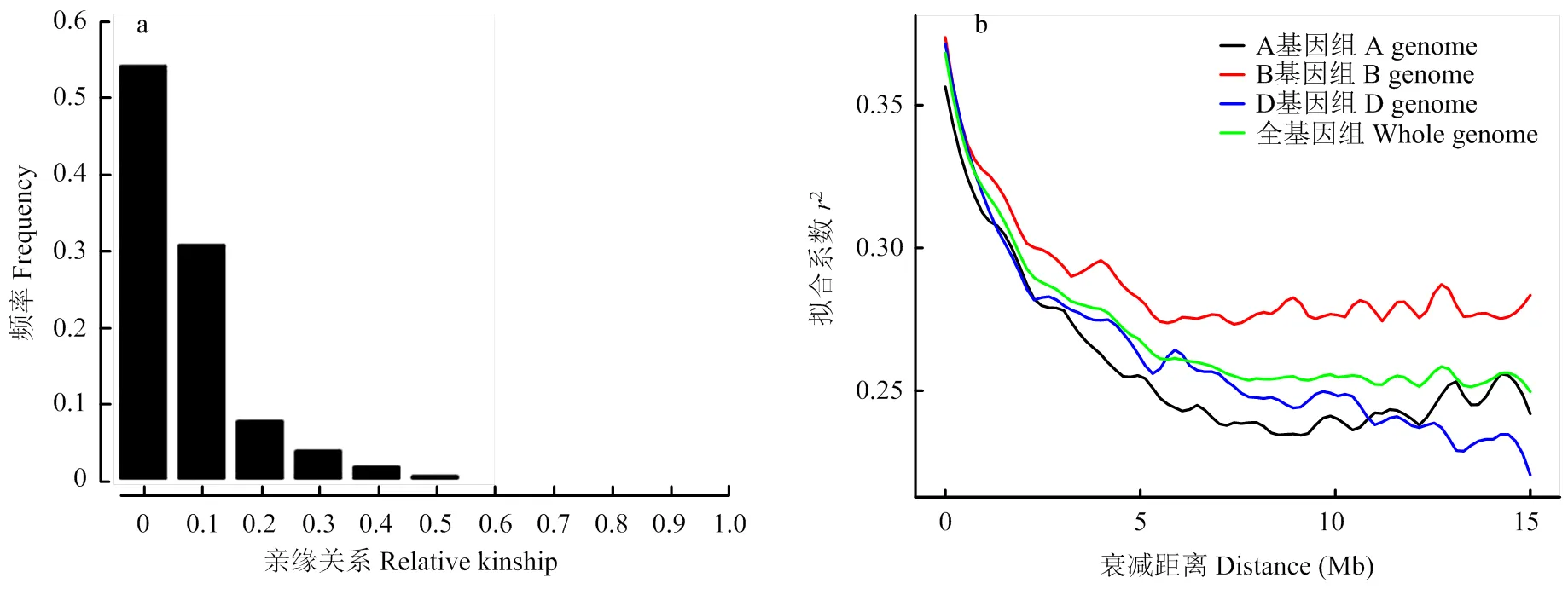
a:品种间相对亲缘关系;b:A、B、D基因组LD衰减图
2.3 全基因组关联分析
为了获得可靠的与目标性状显著关联的SNP位点,同时,利用6个环境下各性状数据及BLUP数据进行关联分析。利用6个环境下各性状数据,9个性状共检测到1 309个显著性SNP标记,其中,每个显著性SNP位点解释的表型变异(phenotypic variation explained,)波动范围为7.259%—70.792%。利用BLUP数据,9个性状共检测到66个显著性位点,同时与2个性状关联的共有SNP有5个,贡献率波动范围为8.498%—21.877%(表1和图3)。
株高(plant height,PH):6个环境下共检测到76个与株高显著关联的SNP位点,分布于除4D以外的其他20条染色体上,单个位点可解释8.191%—29.767%的株高表型变异。上述显著性关联位点中有3个位点同时在2个环境及BLUP值关联结果中被检测到,分别是2A上的AX-108794050(为10.369%,影响效应未知)、2A上的AX-109857508(为11.109%,正效应)、7B上的AX-110969164(为12.632%,正效应);另有2个单一环境位点是与BLUP值关联结果共有的位点,分别是4B上的AX-108838330(为11.109%,影响效应未知)和6A上的AX-109467555(为11.109%,正效应)。利用BLUP值关联分析得到的与株高显著关联的SNP位点有12个,分布在2A、3A、5A、6A、1B、3B、4B、5B和7B上,可解释1.029%—10.143%的株高表型变异,其中2A、6A和5B上各分布了2个;12个显著关联SNP位点中有5个已知正效应的关联SNP位点,效应值为5.771—8.701,其中位于5A的AX-110101561(为10.774%,影响效应未知)的加性效应值最大,为8.701。
穗长(spike length,SL):6个环境下共检测到74个与穗长显著关联的SNP位点,分布于小麦21条染色体上,单个位点可解释8.709%—70.792%的穗长表型变异。上述显著性关联位点均仅在单一环境或数据集中被检测到。其中有10个单一环境位点是与BLUP值关联结果共有的关联位点,分布在1A、2A、5A、6A、3B、7B和2D上,位于1A的AX-111524633解释的表型变异率最大,为36.908%,且为负效应。BLUP值与穗长显著关联的SNP位点有22个,分布在1A、2A、3A、5A、6A、3B、5B、7B、2D、4D、5D和7D上,可解释9.142%—21.877%的穗长表型变异;其中1A、5A、6A和4D上各有3个关联SNP位点,2A、6A和5B上各有2个关联SNP位点;有7个关联位点在6个环境下均未被检测到,已知具有正效应的7个SNP位点的效应值为0.043—0.207。
小穗数(spikelet number,SN):6个环境下共检测到157个与小穗数显著关联的SNP位点,分布于小麦21条染色体上,单个位点可解释8.745%—31.295%的小穗数表型变异,平均为14.528%。157个显著性关联位点中有10个关联位点是与BLUP值关联结果共有的位点,其中9个是单一环境位点,位于3A的AX-108977066(为13.313,负效应)是在3个环境下检测到的重复位点,而位于4D的AX-110123746的正效应值最大,为3.110。BLUP值与小穗数显著关联的SNP位点有12个,分布在1A、3A、4A、5A、7A、6B、7B和4D上,解释9.220%—16.138%的小穗数表型变异,其中3A上有3个,4A和7A上各有2个,其余染色体都仅有1个;有6个已知具有正效应,效应值为0.793—3.110,位于4D上的AX-110123746(为11.129)的正效应值最大,为1.609,位于7A的AX-110515818(为10.816,正效应)在6个环境下均未检测到。
可育小穗数(fertile spikelet number per spike,FSN):6个环境下共检测到209个与可育小穗数显著关联的SNP位点,分布于小麦21条染色体,单个位点可解释8.971%—31.127%的可育小穗数表型变异。其中,有3个关联位点同时在2个环境下及BLUP值关联结果中被检测到,分别是位于1A的AX-111523929(为22.522,影响效应未知)、4D的AX-110123746(为17.663,正效应)及7A的AX-111073226(为12.027,正效应),另有3个单一环境位点在BLUP值关联结果中也被检测到,分别是位于2A的AX-109545508(为16.738,影响效应未知)、6B的AX-111074553(为16.594,影响效应未知)、7B的AX-111700650(为15.936,影响效应未知)。BLUP值与可育小穗数显著关联的SNP位点有7个,分布在1A、2A、7A、4B、6B、7B和4D上,解释11.009%—16.598%的可育小穗数表型变异,其中有3个已知影响效应的位点,位于4D的AX-110123746(为13.514,影响效应未知)的正效应值最大,为1.297;位于4B的AX-111180671(为11.009,正效应)在6个环境下均未检测到。
穗粒数(grain number per spike,GNPS):6个环境下共检测到264个与穗粒数显著性关联的SNP位点,分布于小麦21条染色体,单个位点可解释8.972%—55.210%的穗粒数表型变异,有2个单一环境位点和2个多环境位点同时在BLUP值关联结果中被检测到。2个单一环境位点分别是位于4B的AX-112290916(为16.722,负效应),位于4D的AX-110270363(为21.613,影响效应未知)。2个多环境重复位点分别是位于1A的AX- 108745367(为25.771,影响效应未知),位于6A的AX-110509054(为22.688,负效应)。BLUP值与穗粒数显著关联的SNP位点有8个,可解释8.972%—12.915%的穗粒数表型变异,其中1个位点的染色体位置未知,另7个分别是位于1A的AX-108745367、5A的AX-109470031、6A的AX- 110509054、1B的AX-108786643、4B的AX-112290916、4D的AX-110270363和6D的AX-110509054; 8个位点中已知影响效应的仅有3个,且均为负效应。
千粒重(thousand kernel weight,TKW):6个环境下显著关联到27个千粒重SNP位点,分布在小麦的4A、1B、3B、5B、6B、7B、1D和4D染色体,可解释7.259%—13.687%千粒重表型变异;27个位点均仅在单一环境中被检测到,仅位于3B的AX-110399975(为11.608,负效应)与BLUP值关联位点重复。BLUP值与千粒重显著关联的SNP位点有9个,分布在4A、1B、3B、6B、1D和6D上,解释8.498%—13.687%的千粒重表型变异;其中有6个已知影响效应的位点,位于4A的AX-110401690(为11.570)、6B的AX-109825047(为11.479)和1D的AX-108818349(为12.598)为正效应,位于6B的AX-109825047的效应值最大,为1.299。
粒长(grain length,GL):6个环境下显著关联到438个粒长SNP位点,在小麦21条染色体上均有分布,可解释9.736%—17.537%的粒长表型变异,均为单一环境下检测到的关联位点,其中427个SNP位点是在YN2017这个环境检测到的,占关联位点总数的97.488%;已知位于7B的AX-109477546的正效应值最大,为1.175。
粒宽(grain width,GW):6个环境下显著关联到32个粒宽SNP位点,均为单一环境的独立位点,解释8.690%—15.910%的粒宽表型变异,分布在小麦除1A、1D、3D、4D之外的其他17条染色体上,已知位于6D的AX-110382864正效应值最大,仅为0.145。
籽粒长/宽(grain length/width ratio,GL/GW):6个环境下显著关联到32个籽粒长/宽SNP位点,解释9.232%—49.896%的籽粒长/宽表型变异,均为单一环境下的独立位点,其中28个SNP位点是在YN2017环境下检测到的,占关联位点总数的87.500%。利用BLUP值未检测到与粒长、粒宽、籽粒长/宽显著关联的SNP位点,说明在6个环境下籽粒性状的关联位点具有假阳性。
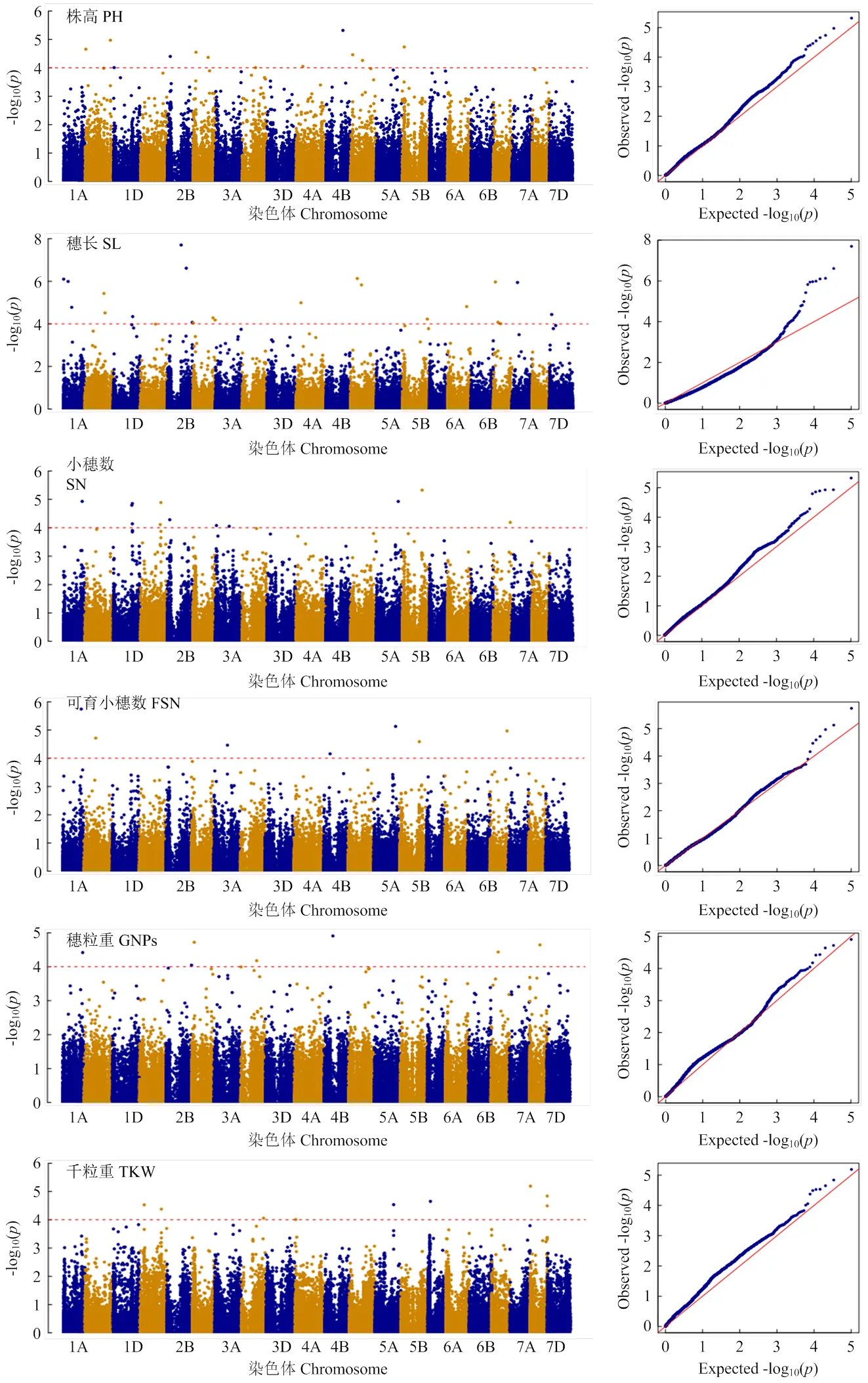
a:株高;b:穗长;c:小穗数;d:可育小穗数;e:穗粒数;f:千粒重 a: Plant height; b: Spike length; c: Spikelet number; d: Grain number per spike; e: Fertile spikelet number per spike; f: Thousand kernel weight
2.4 多性状重复关联位点
在6个环境下检测到的所有显著性关联位点中,仅有2个SNP位点是2个以上性状共有的重复位点,这2个SNP位点利用BLUP值也同样被检测到。一个是位于1A的AX-111523929,分别解释小穗数、可育小穗数、穗粒数的为13.458%、16.598%和23.810%,另一个是位于4D的AX-110123746,分别解释小穗数、可育小穗数、穗粒数的为11.129%、13.514%和22.730%。利用BLUP值检测到的同时与2个以上性状显著关联的SNP重复位点有5个,可解释10.817%—16.598%的表型变异,其中2个SNP位点是小穗数、可育小穗数、穗粒数3个性状的共有位点,即1A的AX-111523929和4D的AX-110123746,另3个SNP位点为小穗数和可育小穗数的共有位点,分别是位于6B的AX-111074553、7A的AX-111073226和7B的AX-111700650;5个位点中位于7A的AX-111073226对小穗数和可育小穗数的正效应值分别为0.794和0.614,位于4D的AX-110123746对小穗数、可育小穗数和穗粒数的正效应值分别为1.609、1.297和18.948,另3个重复位点的影响效应未知。
将同时与2个性状或2个以上环境(BLUP值作为1个环境)相关联的重复SNP位点作为稳定的显著性关联位点,9个性状共计有38个稳定关联SNP位点,包括株高重复位点5个,穗长重复位点10个,小穗数重复位点10个,可育小穗数重复位点6个,穗粒数重复位点6个,千粒重重复位点1个,此38个BLUP值稳定关联SNP位点可以解释9.10%—23.81%的表型变异(表1)。
2.5 关联位点的验证
对群体中的候选重复SNP位点进行多态性验证,不同环境下各性状根据多态性分组的表型值差异均达到极显著水平(<0.01)。基于候选重复SNP位点的成对性状,进行了品种数量汇总和平均值计算(表3)。可以看出,2组成对性状的品种数分别为1—61和116—179,这与6个环境下新疆冬小麦地方品种和育成品种的性状表现基本一致,即134个地方品种的株高、穗长、小穗数、结实小穗数、籽粒长宽比均大于54个育成品种,而穗粒数、千粒重、粒长、粒宽均小于育成品种,育成品种9个农艺产量性状的变异系数均高于地方品种[24]。对单个性状相关的重复SNP进行验证,发现关联到的优良等位基因具有降低株高,增加穗长、小穗数、可育小穗数、穗粒数和千粒重的作用,如AX-108838330中含有等位基因GG的品种有降低株高的作用,AX-110956005中含有等位基因TT的品种有增加穗长的作用,AX-110270363中含有等位基因GG的品种有增加穗粒数的作用(平均为51.39粒),AX-110399975中含有等位基因GG的品种有增加千粒重的作用(平均为35.5 g)。基于与2个以上性状相关的多效性SNP进行验证,如AX-111073226中含有等位基因TT的品种、AX-111523929中含有等位基因AA的品种、AX-111074553中含有等位基因GG的品种、AX-110123746中含有等位基因CC的品种、AX-111700650中含有等位基因CC的品种显示具有更多的小穗数和可育小穗数。
将38个重复SNP位点与已发布小麦基因组(IWGSC RefSeq v1.0:https://urgi.versailles.inra.fr/ download/iwgsc/IWGSC_RefSeq_Annotations/v1.0/)(IWGSC 2018)位点进行比对,仅找到3个与本研究关联重复位点位置相近、且有注释基因功能的基因,一个是2A染色体与本研究株高关联位点AX- 108794050距离相近的,该基因与转录因子bHLH71的代谢合成有关;另一个是1A染色体与本研究穗长关联位点AX-110689765距离相近的,该基因与蛋白质编码有关;4B染色体上与本研究千粒重关联位点AX- 110399975距离相近的,与编码丝氨酸/苏氨酸蛋白激酶SD1-8有关,参与对细胞增殖与分化的调控。
3 讨论
3.1 小麦农艺产量性状表型变异
小麦(L.)是我国第三大粮食作物,对保障粮食供需平衡和国家粮食安全起着至关重要的作用[1]。农艺与产量性状可以直观地反映品种的优劣,具有易观察、好测量的特点。20世纪中叶以来,针对农艺和产量性状的研究在小麦改良中发挥了重要作用。即使在分子标记技术迅猛发展的今天,对小麦种质资源和品种农艺性状的考察、分类与科学评价依然是育种工作的一项重要内容[25]。随着生产条件的不断改善和育种水平的不断提高,我国北部冬麦区小麦品种的组成和主要性状发生了很大变化,主要表现在株高和穗下节由长变短,主穗粒数、穗粒重和千粒重由小变大。这与多年来小麦品种选育以主攻产量的育种目标密切相关[26-27]。马艳明等[28]研究表明,新疆冬小麦育成品种的株高、穗长、小穗数、可育小穗数、穗粒数、籽粒长宽比的变异系数均大于地方品种,而粒长、粒宽的变异系数小于地方品种,说明育成品种的产量性状变异高于地方品种。由于不同品种都有其自身固有的生态适应性,其性状表现是自身遗传因子与环境条件相互作用的结果,而小麦大多数农艺性状(株高、穗长、穗粒数等)均为数量性状,更易受到外界环境条件的影响,这就造成了不同生态条件下种植的小麦品种其生长发育特性差异明显。因此,在不同研究中检测到的QTL在定位位置、QTL数目和遗传效应等方面存在较大差异,给相关研究带来了一定的困难[8, 29-30]。对材料进行多年多点试验可以最大程度上降低生态环境对不同品种农艺产量性状的影响,增加QTL检测的准确性,同时还能对多个农艺性状同时进行关联作图。
表1 38个重复位点的物理位置、值及解释的表型变异率
Fig. 1 Physical locations,values and interpreted phenotypic variation rates of 38 repeat sites
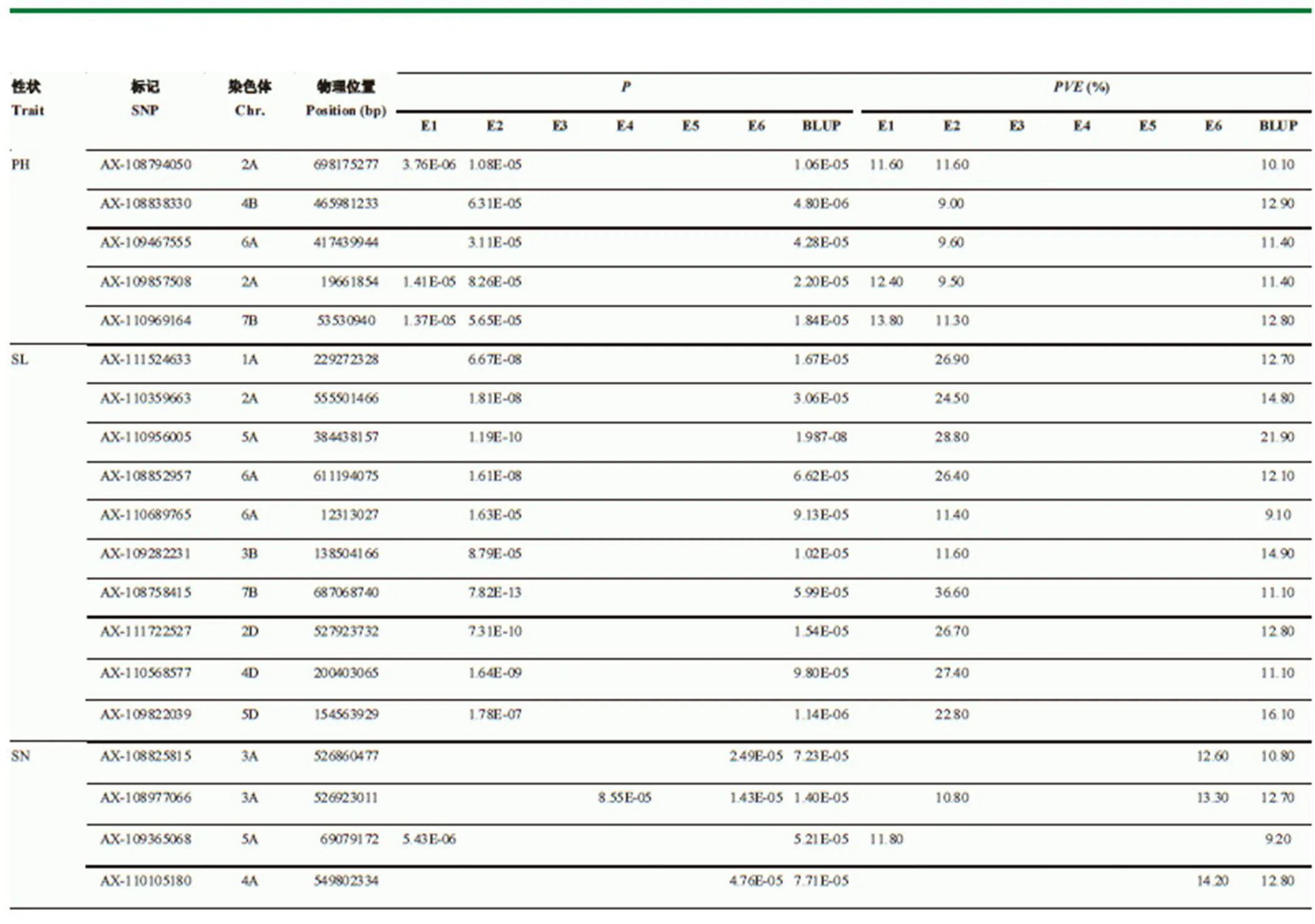
续表1 Continued table 1
PH:株高;SL:穗长;SN:小穗数;FSN:可育小穗数;GNPS:穗粒数;TKW:千粒重;GL:粒长;GW:粒宽;GL/GW:籽粒长/宽
PH: plant height; SL: spike length; SN: spikelet number; FSN: fertile spikelet number per spike; GNPS: grain number per spike; TKW: thousand kernel weight; GL: grain length; GW: grain width; GL/GW: grain length/width ratio
3.2 影响关联分析结果的因素
全基因组关联研究(GWAS)是剖析复杂性状遗传基础的主要方法之一[31],通过利用表型性状和基因本身或基因附近微小区域的分子标记的关联来实现基因的精细定位[31-32]。全基因组关联分析是以生物体在进化过程中的基因重组和基因突变(位点变异)为基础,定位的精细度与统计效力均会受位点数量与样本数量的影响[33]。连锁不平衡(LD)是GWAS的基础,受到遗传衰减、种群分层和自然选择的影响,而群体分层被认为是影响GWAS结果有效性的主要因素[34]。影响LD的因素都会对关联分析的结果产生影响,首要因素就是作物的群体结构。许多重要作物都拥有漫长的驯化史,复杂的选育过程以及来自野生近缘种的遗传漂变,造成了种质资源内存在着复杂的群体结构[22]。当研究所使用的群体存在较多亚群时,等位基因在基因组上的分布往往不平衡,可能造成标记与数量性状相关位点的假阳性关联,从而使关联分析更加复杂[35]。本研究在LD分析的基础上,利用主成分分析法将所有小麦材料分为地方品种和选育品种两个亚群,有效降低了对关联分析结果的影响。
3.3 单个位点对多个性状的遗传效应分析
遗传学上通常会出现一个位点对多个性状产生效应,在作物研究中有很多这样的报道。如Mora等[30]以来自智利、乌拉圭和国际玉米小麦改良中心(CMMITE)的382个春麦品种为材料,采用全基因组扫描法将SNP标记对在不同环境下的株高、穗粒数、千粒重、产量等性状进行关联分析,发现位于2D、1B、3B染色体的iniaGBS30112、iniaGBS33956、iniaGBS36569标记分别只在1个环境下被检测到与产量关联,还有一些标记与多个性状关联。Li等[36]选择326 570个单核苷酸多态性(SNP)标记对来自中国黄淮麦区的166个优质小麦品种进行9个产量相关性状关联分析,在8条染色体上检测到12个多效性基因位点;其中3A染色体上的714.4—725.8 Mb区间与产量、穗粒数、粒宽、株高、最长节间长度和旗叶长显著相关。Sun等[8]利用90K SNP芯片对163个小麦品种的13个产量相关性状进行关联分析,检测到5个多效性SNP与3个或更多性状相关。本研究检测到5个多效SNP同时与小穗数、结实小穗数显著关联,而小穗数与结实小穗数之间存在极显著相关性,因此必然会有相同的显著关联位点。
4 结论
利用多位点全基因组关联分析方法(MLM)对小麦9个产量相关性状进行了全基因组关联分析,在6个环境和综合BLUP值条件下分别检测到1 309和66个与性状显著关联的SNP位点,其中,与2个性状或2个以上环境(BLUP值作为1个环境)相关联的重复SNP位点有38个。结合基因功能注释,筛选出3个重要的产量相关性状候选基因。
[1] 何中虎, 庄巧生, 程顺和, 于振文, 赵振东, 刘旭. 中国小麦产业发展与科技进步. 农学学报, 2018, 8(1): 99-106.
He Z H, Zhuang Q S, Cheng S H, Yu Z W, Zhao Z D, Liu X. Wheat production and technology improvement in China. Journal of Agriculture, 2018, 8(1): 99-106.(in Chinese)
[2] 吴澎, 刘娟, 田纪春. 单核苷酸多态性(SNP)分子标记在小麦遗传育种中的研究进展. 农学学报, 2019, 9(1): 54-58.
Wu P, Liu J, Tian J C. Research progress of single nucleotide polymorphism (SNP) molecular markers in wheat genetic breeding. Journal of Agriculture, 2019, 9(1): 54-58. (in Chinese)
[3] Huang X H, Han B. Natural variations and genome-wide association studies in crop plants. Annual Review of Plant Biology, 2014, 65: 531-551.
[4] Cao S H, Xu D A, Hanif M, Xia X C, He Z H. Genetic architecture underpinning yield component traits in wheat. Theoretical and Applied Genetics, 2020, 133(6): 1811-1823.
[5] Qaseem M F, Qureshi R, Muqaddasi Q H, Shaheen H, Kousar R, RÖder M S. Genome-wide association mapping in bread wheat subjected to independent and combined high temperature and drought stress. PLoS One, 2018, 13(6): e0199121.
[6] SUKUMARAN S, DREISIGACKER S, LOPES M, CHAVEZ P, REYNOLDS M P. Genome-wide association study for grain yield and related traits in an elite spring wheat population grown in temperate irrigated environments. Theoretical and Applied Genetics,2015, 128(2): 353-363.
[7] Chen G F, Zhang H, Deng Z Y, Wu R G, Li D M, Wang M Y, Tian J C. Genome-wide association study for kernel weight-related traits using SNPs in a Chinese winter wheat population.Euphytica, 2016, 212(2): 173-185.
[8] Sun C W, Zhang F Y, Yan X F, Zhang X F, Dong Z D, Cui D Q, Chen F. Genome-wide association study for 13 agronomic traits reveals distribution of superior alleles in bread wheat from the Yellow and Huai Valley of China. Plant biotechnology journal, 2017, 15(8): 953-969.
[9] Wang S X, Zhu Y L, Zhang D X, Shao H, Liu P, Hu J B, Zhang H, Zhang H P, Chang C, Lu J, Xia X C, Sun G L, Ma C X. Genome-wide association study for grain yield and related traits in elite wheat varieties and advanced lines using SNP markers. PLoS One, 2017, 12(11): e0188662.
[10] Ye X L, Li J, Cheng Y K, Yao F J, Long L, Wang Y Q, Wu Y, Li J, Wang J R, Jiang Q T, Kang H Y, Li W, Qi P F, Lan X J, Ma J, Liu Y X, Jiang Y F, Wei Y M, Chen X M, Liu C J, Zheng Y L, Chen G Y. Genome-wide association study reveals new loci for yield-related traits in Sichuan wheat germplasm under stripe rust stress. BMC genomics, 2019, 20(1): 640.
[11] 翟俊鹏, 李海霞, 毕惠惠, 周思远, 罗肖艳, 陈树林, 程西永, 许海霞. 普通小麦主要农艺性状的全基因组关联分析. 作物学报, 2019, 45(10): 1488-1502.
Zhai J P, Li H X, Bi H H, Zhou S Y, Luo X Y, Chen S L, Cheng X Y, Xu H X. Genome-wide association study for main agronomic traits in common wheat. Acta Agronomica Sinica, 2019, 45(10): 1488-1502. (in Chinese)
[12] 赵丹阳, 朱婷, 王卫东, 张思妮, 夏雪姣, 翟晓光, 丁勤, 马翎健. 黄淮麦区部分小麦品种(系)重要产量性状全基因组关联分析. 麦类作物学报, 2018, 38(11): 1320-1329.
Zhao D Y, Zhu T, Wang W D, Zhang S N, Xia X J, Zhai X G, Ding Q, Ma L J. Genome-wide association study of yield traits in some wheat varieties (lines) of Huang-Huai area. Journal of Triticeae Crops, 2018, 38(11): 1320-1329. (in Chinese)
[13] 顾晶晶, 余慷, 陈树林, 朱保磊, 王冬至, 张爱民, 刘冬成, 詹克慧. 河南小麦品种穗粒数性状的动态变化及全基因组关联分析. 分子植物育种, 2017, 15(10): 4143-4158.
Gu J J, Yu K, Chen S L, Zhu B L, Wang D Z, Zhang A M, Liu D C, Zhan K H. Dynamic variation and genome-wide association analysis of grain number related traits in Henan wheat (). Molecular Plant Breeding, 2017, 15(10): 4143-4158. (in Chinese)
[14] 马艳明, 娄鸿耀, 陈朝燕, 肖菁, 徐麟, 倪中福, 刘杰. 新疆冬小麦地方品种与育成品种基于SNP芯片的遗传多样性分析. 作物学报, 2020, 46(10): 1539-1556.
Ma Y M, Lou H Y, Chen Z Y, Xiao J, Xu L, Ni Z F, Liu J.Genetic diversity assessment of winter wheat landraces and cultivars in Xinjiang via SNP array analysis.Acta Agronomica Sinica2020, 46(10): 1539-1556. (in Chinese)
[15] 李立会, 李秀全, 杨欣明. 小麦种质资源描述规范和数据标准. 北京: 中国农业出版社, 2006.
Li L H, Li X Q, YANG X M. Descriptors and data standard for wheat (L.). Beijing: China Agriculture Press, 2006. (in Chinese)
[16] Saghai-Maroof M A, Soliman K M, Jorgensen R A, Allard R W. Ribosomal DNA spacer-length polymorphisms in barley: mendelian inheritance, chromosomal location, and population dynamics. Proceedings of the National Academy of Sciences of the United States of America, 1984, 81(24): 8014-8018.
[17] Pritchard J K, StephenS M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics, 2000, 155(2): 945-959.
[18] Evanno G, Regnaut S, Goudet J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Molecular ecology, 2005, 14(8): 2611-2620.
[19] Jakobsson M, Rosenberg N A. CLUMPP: A cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics, 2007, 23(14): 1801-1806.
[20] Yu J M, Buckler E S. Genetic association mapping and genome organization of maize. Current Opinion in Biotechnology, 2006, 17(2): 155-160.
[21] Audic S, Claverie J M. The significance of digital gene expression profiles. Genome Research, 1997, 7(10): 986-995.
[22] YANG N, LU Y L, YANG X H, HUANG J, ZHOU Y, ALI F, WEN W W, LIU J, LI J S, YAN J B. Genome wide association studies using a new nonparametric model reveal the genetic architecture of 17 agronomic traits in an enlarged maize association panel. PLoS Genetics, 2014, 10(9): e1004573.
[23] Hardy O J, Vekemans X. Spagedi: A versatile computer program to analyse spatial genetic structure at the individual or population levels. Molecular Ecology Notes, 2002, 2(4): 618-620.
[24] STICH B, MELCHINGER A E, FRISCH M, Maurer Hans P, Heckenberger M, Reif J C. Linkage disequilibrium in European elite maize germplasm investigated with SSRs. Theoretical & Applied Genetics, 2005, 111(4): 723-730.
[25] 董玉琛, 郑殿升. 中国小麦遗传资源. 北京: 中国农业出版社, 2000.
Dong Y C, Zheng D S. Chinese wheat genetic resources. Beijing: China Agriculture Press, 2000. (in Chinese)
[26] 刘三才, 郑殿升, 曹永生, 宋春华, 陈梦英. 中国小麦选育品种与地方品种的遗传多样性中国农业科学, 2000, 33(4): 20-24.
Liu S C, Zheng D S, Cao Y S, Song C H, Chen M Y. Genetic diversity of landrace and bred varieties of wheat in China. Scientia Agricultura Sinica, 2000, 33(4): 20-24. (in Chinese)
[27] 李煦征. 江苏省小麦品种更替过程中生态型演化与区域差异分析[D]. 南京: 南京农业大学, 2007.
LI X Z. Research on the evolution of ecotypes of wheat cultvars and differences of regions in Jiangsu[D]. Nanjing: Nanjing Agricultural University, 2007. (in Chinese)
[28] 马艳明, 冯智宇, 王威, 张胜军, 郭营, 倪中福, 刘杰. 新疆冬小麦品种农艺及产量性状遗传多样性分析. 作物学报, 2020, 46(12): 1997-2007.
MA Y M, FENG Z y, WANG W, ZHANG S J, GUO Y, NI Z F, LIU J. Genetic diversity analysis of winter wheat landraces and cultivars in Xinjiang based on agronomic traits.Acta Agronomica Sinica, 2020, 46(12): 1997-2007. (in Chinese)
[29] Risch N, Merikangas K. The future of genetic studies of complex human diseases.Science, 1996, 273(5281): 1516-1517.
[30] Mora F, Castillo D, Lado B, Matus I, Poland J, Belzile F, von Zitzewitz J, del Pozo A. Genome-wide association mapping of agronomic traits and carbon isotope discrimination in a worldwide germplasm collection of spring wheat using SNP markers. Molecular Breeding, 2015, 35(2): 69.
[31] INGVARSSON P K, NATHANIEL R S. Association genetics of complex traits in plants. New Phytologist, 2011, 189: 909-922.
[32] Zhu C, Gore M, Buckler E S, Yu J. Status and prospects of asso-ciation mapping in plants. Plant Genome, 2008, 1: 5-20.
[33] 易腾飞. 中国冬麦区小麦品种农艺性状与品质性状的全基因组关联分析[D]. 保定: 河北农业大学, 2018.
Yi T F. Genome-wide association study of agronomic traits and quality traits of wheat varieties in the winter wheat region of China[D]. Baoding: Hebei Agricultural University, 2018. (in Chinese)
[34] Cardon L R, Palmer L J. Population stratification and spurious allelic association. Lancet, 2003, 361(9357): 598-604.
[35] 郭玉华, 朱四光, 张龙步, 都华. 不同栽培条件对水稻茎秆材料学特性的影响. 沈阳农业大学学报, 2003, 34(1): 4-7.
GUO Y H, ZHU S G, ZHANG L B, DU H. Influence on the material characteristics of rice culms in different cultivation conditions. Journal of Shenyang Agricultural University, 2003, 34(1): 4-7. (in Chinese)
[36] Li F J, Wen W E, Liu J D, Zhang Y, Cao S H, He Z H, Rasheed A, Jin H, Zhang C, Yan J, Zhang P Z, Wan Y X, Xia X C. Genetic architecture of grain yield in bread wheat based on genome‒wide association studies. BMC Plant Biology, 2019, 19(1): 1-19.
Genome-wide association analysis of Yield Traits in Xinjiang Winter Wheat Germplasm

1Institute of Crop Germplasm Resource, Xinjiang Academy of Agricultural Sciences, Urumqi 830091;2College of Agronomy and Biotechnology, China Agricultural University, Beijing 100193;3Institute of Hybrid Wheat, Beijing Academy of Agriculture and Forestry Sciences, Beijing 100097;4Institute of Agricultural Sciences of Ili Prefecture, Yining 835011, Xinjiang;5College of Agronomy, Shandong Agricultural University, Tai’an 271018, Shandong
【Objective】To discover new high yield genes in wheat by association analysis, which can provide technical supports for the innovation and genetic improvement of high yield germplasm resources in wheat.【Method】Totally 188 bread wheat cultivars in Xinjiang were genotyped using the wheat 55K genotyping assay. GWAS was carried out to identify the signifcant single nucleotide polymorphisms (SNPs) which were associated with 9 wheat yield traits in 6 environments. The MLM algorithm in TASSEL5.0 was used to analyze the nine traits related to wheat yield traits.【Result】Totally 1309 SNPs explained 7.259%-70.792% of the phenotypic variation. 38 SNP loci were identifed, which were significantly correlated with 5 plant height weight SNP loci, 10 spike length weight SNP loci, 10 spikelet number SNP loci, 6 fertile spikelet number SNP loci, 6 spike grain number SNP loci, and 1 thousand grain weight SNP loci. These loci can explain 9.10%-23.81% of phenotypic variations. Comparing these 38 loci with the published wheat genome loci, only 3 functional genes were found, which annotated with gene function. There genes are:on chromosome 2A, which is close to the plant height associated site AX-108794050 and is related to the metabolic synthesis of transcription factor bHLH71;, located on chromosome 1A at a distance similar to the spike length associated site AX-110689765, is related to protein coding;, located on the 4B chromosome at a distance similar to the 1000 grain weight associated site AX-110399975, is associated with the encoding serine/threonine protein kinase SD1-8 and is involved in regulating cell proliferation and differentiation. 【Conclusion】38 QTL loci associated with wheat yield traits were detected. After verification, it was found that the associated excellent alleles have the effect of reducing plant height, increasing spike length, spikelet number, fertile spikelet number, grain number per spike, and thousand grain weight.
wheat; yield traits; SNP; association analysis; candidate genes
10.3864/j.issn.0578-1752.2023.18.001
2023-03-27;
2023-05-25
国家科技资源共享服务平台-国家作物种质资源库项目(NCGRC-2021-029)
马艳明,Tel:13999260539;E-mail:ymma213@sina.com。通信作者刘杰,Tel:010-62734072;E-mail:jieliu@cau.edu.cn
(责任编辑 李莉)
