脱木质素处理对速生杨木和杉木细胞壁层结构影响
2023-10-17孙呵龚翌之燕韵天李珊付常青常旬陈太安
孙呵,龚翌之,燕韵天,李珊,付常青,常旬,陈太安*
(1. 西南林业大学材料科学与工程学院,昆明 650224;2. 厦门大学中国-东盟海洋学院,厦门 361101)
木材是具有多尺度孔隙结构的复杂毛细管系统,木材相互连通的细胞腔及纹孔构成了大毛细管系统,细胞壁内相互连通的微毛细管构成了微毛细管系统。针叶材大毛细管系统中的管胞和纹孔口直径分别为10~50 μm和0.02~4 μm,阔叶材大毛细管系统中导管直径为20~400 μm,而纹孔开口略小于针叶材[1],因此,大毛细管系统中组织的孔径多为微米级。而微毛细管系统主要在细胞壁中,其多由微纤丝间和纤丝间的孔隙组成,孔径多在介孔(2~50 nm)和微孔(<2 nm)范围,其中介孔又以2~10 nm为主。Shi等[2]研究发现,木材细胞壁中2 nm以下的微孔体积数倍于介孔体积。Nakatani等[3]研究发现,木质素中存在大量0.6 nm以下的孔隙,而纤维素和半纤维素中几乎没有发现此类孔隙。此外,大毛细管系统对水分束缚力很小,是自由水主要存在的地方,微毛细管系统内的微纳孔隙是结合水存在的主要位置,因此,无论是自由水还是结合水,均与木材孔隙结构密切相关。
不同相对湿度下,水分子进入细胞壁的机理并不一致。中低湿度(相对湿度<98%)下的吸湿性主要是木材中活性羟基等亲水性基团以范德瓦耳斯力与空气中的水蒸气结合形成氢键,进而生成单分子与多分子层水,因此,这部分水分主要与半纤维素和无定型区的纤维素活性羟基有关。然而,高相对湿度(相对湿度>98%)下的吸湿性与中低湿度下有较大不同,主要是由于大毛细管和微毛细管系统具有较高的孔隙率和内表面积。当内部的饱和蒸汽分压小于环境中的饱和蒸汽分压时,将会产生毛细管凝缩现象,且会在较小的高湿度范围显著提升含水率,但其仍属于吸湿性范畴,因此,高湿度下吸湿性以不同孔径范围的毛细管凝缩作用为主,与孔径直接相关[4]。此外,中低湿度和高湿度下的吸湿是逐渐过渡的,没有明显的界限[5],不同湿度下的单分子层吸附、多分子层吸附和毛细管凝缩等机理在各个湿度下共同作用进而提升吸湿性。在中低湿度下,Rautkari等[6]研究表明,热处理材的羟基可及性与平衡含水率关联较差,须有额外的机制来解释吸湿性的变化。Himmel等[7]研究发现,乙酰化处理材在相对湿度20%以下时吸湿性的降低主要是因为活性羟基被覆盖,而在相对湿度20%以上时吸湿性的下降只是由细胞壁的物理膨胀造成的。Thybring等[8]研究认为,化学改性材中水分的变化与羟基可及性无关,细胞壁中微纳孔隙才是木材吸湿性的潜在控制机制。综上所述,木材吸湿性受孔隙结构和吸湿基团等物理和化学因素双重控制,目前,高湿度和中低湿度下木材的吸湿机理均未明晰,特别是细胞壁孔隙结构对于吸湿性的影响有待进一步明确。
基于影响木材吸湿性因素的复杂性,本研究采用杨木与杉木为研究对象,采用不同程度的脱木质素处理,达到调节细胞壁孔隙结构等物理特性与吸湿性基团等化学性质的目的,进而分析吸湿性的变化,明确孔隙结构变化对于吸湿性的影响,以期为木材-水分关系的完善提供参考。
1 材料与方法
1.1 试验材料
市购杨木(Populussp.)、杉木(Cunninghamialanceolata)边材,选择纹理通直、无可见缺陷板材,加工成25 mm(弦向)×10 mm(径向)×5 mm(纵向)的试样若干备用。亚氯酸钠(分析纯)、过氧化氢(质量分数为30%)、冰乙酸(质量分数为98%),天津市风船化学试剂科技有限公司。乙醇(分析纯),四川西陇科学有限公司。蒸馏水为自制。
1.2 试验方法
1)试样预处理。首先配制1%和2%质量分数的亚氯酸钠溶液,用冰乙酸将其pH调至4.6,同时用蒸馏水代替亚氯酸钠溶液做对照处理;之后将溶液与试件置于干燥皿中抽真空60 min,使试件完全沉没,其中,试件与溶液的比例为每个试件40 mL溶液;最后在20 ℃烘箱内静置过夜。
2)脱木质素材制备。将试件与溶液置于接有冷凝装置的锥形瓶中,80 ℃水浴加热8 h,每隔4 h更换一次新鲜溶液,制备出木质素脱出率为24.19% 和32.09%的杉木低强度、高强度脱木质素材,以及13.07%和21.90%的低强度和高强度杨木脱木质素材。杨木对照材、低强度和高强度脱木质素材分别命名为UDP、LDP、HDP,杉木的则分别命名为UDF、LDF、HDF。其中,木质素含量参照文献[9],木质素脱出率为木质素脱出量与处理前绝干样品质量的比值。之后进行泡水处理,去除残余溶液,过程为前3天每8 h换水,之后每12 h换水,直至换水溶液呈中性。最后进入干燥程序,脱木质素材干燥采用乙醇置换的方法,将试件经过质量分数为35%,50%,75%的乙醇溶液各5 d的置换,再经过90%质量分数乙醇4个月置换,最后用100%乙醇置换7 d,然后放入干燥皿使乙醇挥发,质量不变后进行吸湿试验。
3)微观构造观察。采用生物数码显微镜(ECLIPSE 80i型,日本Nikon)中的偏光显微镜分析脱木素后纤维素的分布变化情况,采用荧光显微镜分析脱木素后木质素的分布变化情况,采用普通光(亮场)下的显微图片观察细胞形态变化。观察切片为10~12 μm横切面,制样方法同文献[10]。
4)木材化学基团及组分变化。采用傅里叶变换红外光谱仪(IS50型,美国Nicolet)分析脱木素前后的官能团变化。测试时将100~120目(粒径0.125~0.150 mm)烘干处理的粉末与溴化钾混合后压片进行红外光谱分析,其中两者质量比为1∶200,扫描范围400~4 000 cm-1,扫描次数64次。
5)结晶度分析。采用X射线衍射仪(XRD,Ultima IV型,日本Rigaku)分析脱木素前后的结晶度变化。样品为100~120目(粒径0.125~0.150 mm)烘干处理的粉末,铜靶,Kα辐射,电压40 kV,电流40 mA,10°~90°范围连续扫描,扫描速度为1.8(°)/min,采样间隔0.015 0°。结晶度计算根据Segal经验公式[11]。
6)脱木质素对孔隙结构的影响。采用气体吸附-脱附分析仪(Micromeritics ASAP 2460型)表征脱木质素前后的介孔变化。测试前在80 ℃和低于6.67×10-2Pa的真空度下脱气12 h以上,之后采用全孔分析模式,以氮气作为吸附介质,试验温度为77.4 K。吸附-脱附的相对压力(P/P0)选择在0.01~0.995。样品80 ℃烘干至质量恒定后进行测试。氮气等温吸附-脱附曲线根据密度泛函理论(density function theory,DFT)平衡模型进行计算,孔径分布利用B-J-H(Barrett-Jotner-Halenda)模型计算,进而获得总孔容和平均直径。
采用压汞仪(Micromeritics AutoPore IV型)分析不同强度脱木质素处理后试样中大孔的变化。试验过程中设备自动控制注汞率,压力范围0~414 MPa,进汞时压力从0 MPa阶梯式升高至414 MPa,退汞时从414 MPa阶梯式降至0 MPa,设定低压和高压汞平衡时间均为10 s。样品80 ℃烘干至质量恒定后进行测试。样品中的孔隙直径D根据公式(1)计算:
D=-2γcosθ/P
(1)
式中:γ为测试液体汞的表面张力;θ为汞与细胞壁的接触角;P为测试时施加的压力。
7)吸湿性试验。将乙醇置换后的干燥试件悬挂在20 ℃,相对湿度依次为35%,50%,60%,70%,80%,85%的恒温恒湿箱(KMF 720, Binder company型)中吸湿,在各湿度下平衡后称质量。之后,在箱体中注满饱和蒸汽,使其相对湿度接近100%,样品在该湿度下平衡后再依次在相对湿度85%,80%,70%,60%,50%,35%的环境中解吸,每组试件重复10~12个。恒温恒湿箱温度、相对湿度及天平的精度分别为±0.1 ℃、2.5%和0.000 1 g。吸湿平衡的标准是样品在每个湿度下吸湿192 h后,每隔24 h测试1次,两次测量质量相差0.1%以内视为平衡。吸湿试验结束后样品在103 ℃烘干24 h,最终计算平衡含水率和吸湿滞后。
吸湿与解吸平衡含水率MC-AD和MC-DE采用式(2)计算:
MC=(M2-M1)/M1×100%
(2)
式中:M1为试样吸湿或者解吸前的绝干质量,g;M2为试样吸湿或者解吸平衡质量,g。
吸湿滞后ΔMC采用式(3)计算:
ΔMC=MC-DE-MC-AD
(3)
8)脱木质素处理材Hailwood-Horrobin等温吸附曲线拟合。为进一步解析脱木质素处理对于木材吸湿性的影响,借助在木材科学领域应用较广的Hailwood-Horrobin(H-H)模型进行分析。H-H模型将木材-水分体系视为理想溶液,并认为该体系由3种要素构成,即绝干木材、水合木材和溶解水[12],因此,吸着水也可以分为水合水和溶解水,它们与不同相对湿度的关系可表示为:
(4)
同时可整理为:
(5)
其中:
(6)
(7)
(8)
式中:EMC为平衡含水率;RH为相对湿度;Mh为水合水平衡含水率;Md为溶解水平衡含水率;K1、K2分别为单、多分子层吸附时水分-木材体系的平衡常数;W为含有单位摩尔吸附点的绝干木材质量。
2 结果与分析
2.1 脱木质素处理对微观构造的影响
木材细胞壁由于结晶区纤维素分子的有序排列而具有双折射性,通过偏光显微镜可以观察到处理前后木材细胞壁内纤维素骨架的变化情况。植物细胞壁木质素中酚类物质能产生自发荧光[3],所以通过荧光显微镜可凭自发荧光的强弱来判定植物细胞壁内木质素浓度的高低。
脱木质素处理前后的杉木与杨木的普通光、荧光、偏光下的图片见图1。由图1可以看出,在普通光下(图1a、d、g),脱木质素处理前后的杉木材细胞形态没有明显变化,均呈椭圆形或圆形。在偏光显微镜下(图1c、f、i),脱木质素对于杉木材细胞壁中的纤维素破坏较小,没有改变纤维素在细胞壁的分布规律。在荧光显微图片中(图1b、e、h),UDF的荧光集中在细胞角隅和胞间层,LDF和HDF相比UDF细胞壁次生壁中的荧光现象减弱,可能是由于脱木质素处理将次生壁S3层表面的木质素薄层脱出导致的。同时,LDF中细胞角隅和胞间层中已经出现部分孔隙(图1e),HDF中细胞角隅和胞间层出现大量缝隙(图1h),表明脱木质素处理将细胞角隅和胞间层中的木质素大量脱出。脱木质素改变了木质素在木材微区的原有分布规律,在UDF中木质素浓度从大到小依次为细胞角隅、复合胞间层、次生壁,脱木质素处理后则相反,木质素浓度从大到小依次为次生壁、复合胞间层、细胞角隅。从杨木显微图片(图1j~r)可以看出,UDP中细胞角隅和胞间层中的木质素浓度较高,脱木质素材细胞壁荧光亮度下降,且在细胞角隅和胞间层也产生了明显的缝隙,与杉木基本一致,但杨木细胞角隅和胞间层的孔隙明显小于杉木。

图1 脱木质素处理前后杉木与杨木的普通光、荧光、偏光图Fig. 1 The observation of bright-field light, polarized light and fluorescence of Chinese fir and poplar before and after delignification
2.2 木材主要组分含量变化

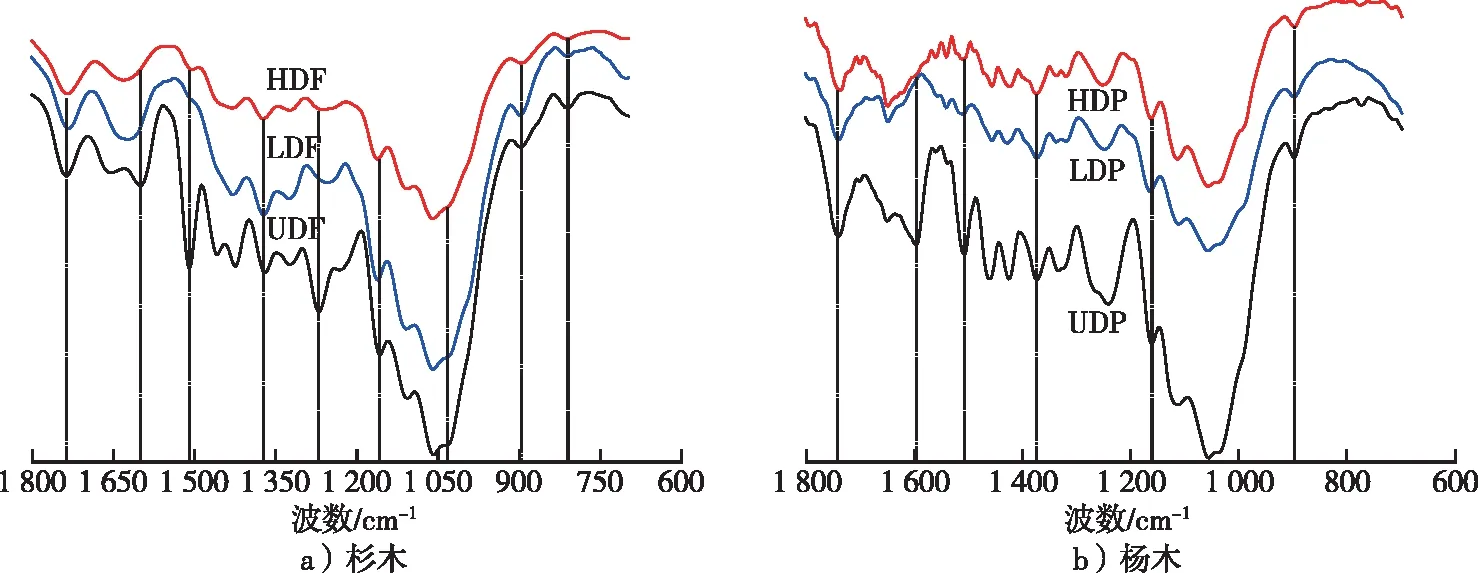
图2 杉木与杨木脱木质素前后的红外光谱Fig. 2 FT-IR of untreated and delignification-treated Chinese fir and poplar
杨木材和杉木材脱木质素前后组分相对含量变化见表1。从表1可以看出,UDF、LDF、HDF中代表木质素特征峰(1 508 cm-1)和代表纤维素、半纤维素特征峰(1 375 和1 157 cm-1)的峰高比值分别从2.48,1.63变化到0.72,0.36和0.20,0.10,表明了木质素不同程度的脱出。杉木脱木质素前后半纤维素的特征峰(1 739 cm-1)和纤维素、半纤维素的特征峰(1 375 和1 157 cm-1)的峰高比值从1.70,0.81变化到2.89,1.19和1.78,0.93,表明脱木质素后半纤维素的相对含量增加,但是HDF的半纤维素相对含量小于LDF。纤维素特征峰(1 375 和1 157 cm-1)和木质素特征峰(1 508 cm-1)的峰高比值表明脱木质素后纤维素的相对含量显著增加。此外,杨木和杉木的纤维素和木质素含量变化一致,而脱木质素材的半纤维素相对于未处理材,其含量显著增加,但高强度脱木质素下的略有差异,这可能与杨木和杉木的半纤维素含量差异有关。
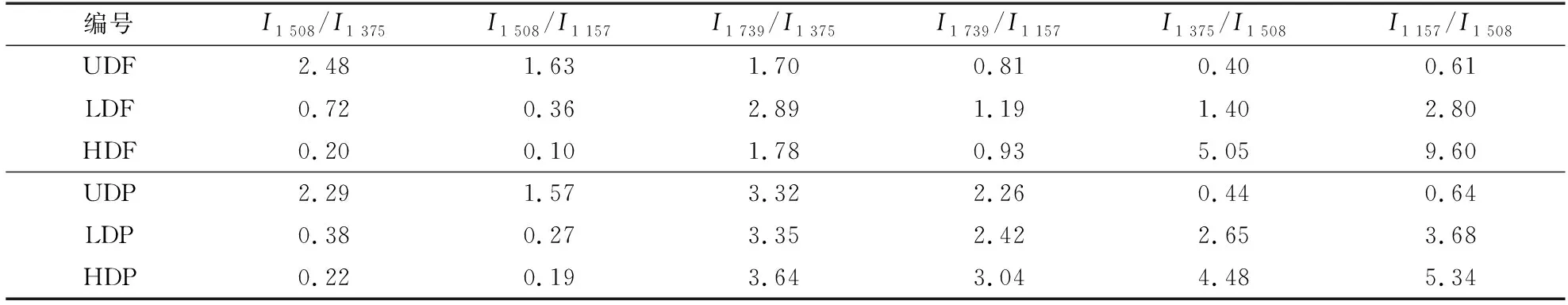
表1 脱木质素处理前后木质素、纤维素、半纤维素的相对峰强度变化Table 1 Changes in relative peak intensity of lignin, cellulose and hemicellulose before and after delignification treatment
2.3 脱木质素对纤维素结晶度的影响
脱木质素处理前后杨木材和杉木材的结晶度变化见图3。从图3可以看出,脱木质素没有改变杨木材和杉木材纤维素Ⅱ型的结晶结构,XRD衍射峰中22°附近002晶面的衍射峰代表结晶区大小,18°附近波谷代表无定型区大小。杨木UDP、LDP、HDP的结晶度分别是40.59%,48.53%,52.02%;杉木UDF、LDF、HDF的结晶度分别为38.39%,44.12%,45.13%。可以看出,脱木质素明显增加了结晶度,与脱木质素后002晶面的衍射峰变化一致,这可能是由于脱木质素处理后纤维素结晶区聚集导致的。不同程度脱木质素处理的杨木材结晶度都明显大于杉木材,说明脱木质素处理没有改变杨木纤维素结晶度大于杉木的本质。

图3 杨木与杉木脱木质素前后的结晶度变化Fig. 3 Crystallinity change of untreated and delignification-treated poplar and Chinese fir
2.4 脱木质素处理对孔隙结构的影响
2.4.1 脱木质素对细胞壁介孔的影响
1)氮气吸附-脱附等温线特征。脱木质素处理前后杨木材与杉木材的吸附-脱附等温线见图4。根据IUPAC(国际纯粹与应用化学联合会)的分类方法,可以判定UDP、LDP吸附-脱附等温线均为Ⅱ和Ⅳ型混合等温线,出现H3型滞后环,表明均存在裂隙状孔隙,不存在明显的墨水瓶孔隙;但是HDP为Ⅳ型吸附-脱附等温线,出现H2型滞后环,表明存在墨水瓶孔隙。从杉木的吸附-脱附等温线可以看出,UDF、LDF、HDF均为Ⅱ和Ⅳ的混合等温线,出现H3型滞后环,且均为裂隙状孔隙,而滞后环随着脱木质素程度的提高而变大。

图4 杨木和杉木脱木质素前后的N2吸附-脱附等温线Fig. 4 Adsorption-desorption isotherm of N2 for Chinese fir and poplar before and after delignification
2)细胞壁介孔分布。脱木质素处理前后的杨木材与杉木材孔径分布及孔容占比见图5,其中孔容占比根据孔径分布中最明显变化的2~10 nm、较明显变化的10~30 nm及不明显变化的30~100 nm 3个区段进行分类计算。可以看出,UDF和UDP的孔径主要在2~30 nm间分布,并以2~10 nm间居多,而脱木质素处理明显增加了杨木材和杉木材2~30 nm范围内的孔隙,且在8 nm附近孔容增量变化最为明显,与之前的研究基本一致[13]。UDP在2~30 nm内的孔隙占总孔容的56.49%,UDF为63.74%,而高强度脱木质素的HDP为79.21%,HDF为91.31%,此部分介孔的增加可能是在木质素间微纳孔隙的基础上产生的。也有学者发现,脱木质素最先破坏的是木质素间的微孔[3,14]。脱木质素处理对于杉木介孔内的孔隙直径变化范围和孔体积增量的影响均大于杨木,尤其是HDF的介孔增量明显大于HDP,这可能是由于杉木材的木质素脱出量大于杨木材而导致的。相比低强度脱木质素和未处理材,高强度脱木质素的杨木材和杉木材孔隙平均直径减小,主要是因为氮气吸附法中测量的孔径范围多在2~50 nm,脱木质素处理后,杨木材和杉木材2~10 nm间孔径数量的增加导致测试中总孔体积的增加和平均孔直径的下降。
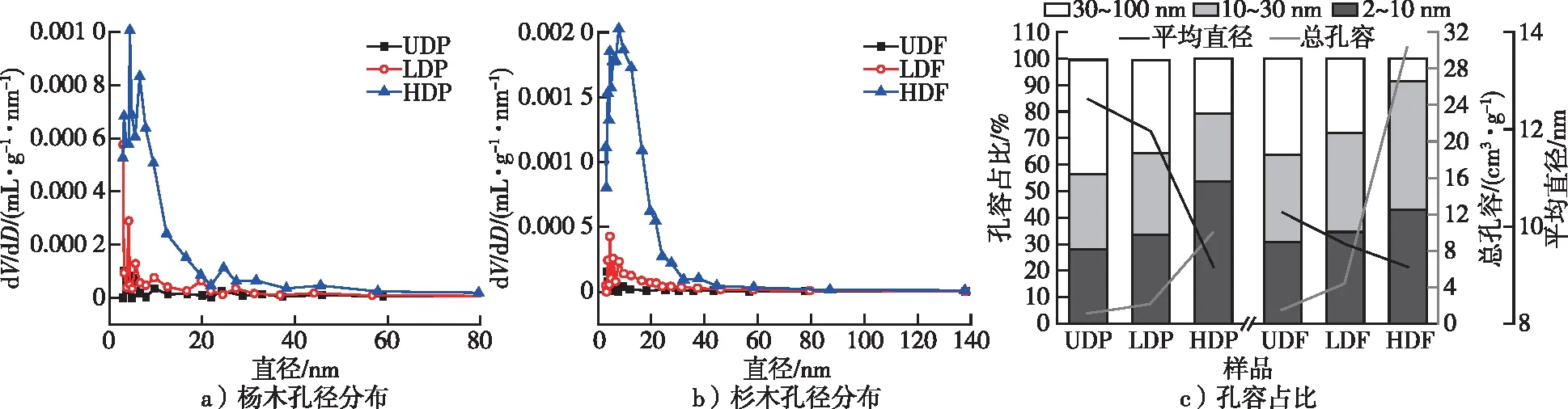
图5 氮气吸附-脱附法分析杨木和杉木脱木质素前后的孔径分布和孔容占比Fig. 5 Analysis of pore size distribution and pore volume ratio of poplar and Chinese fir before and after delignification using nitrogen adsorption-desorption method
2.4.2 脱木质素对细胞壁中大孔的影响
脱木质素处理前后杨木材和杉木材的累积孔体积、微分孔体积及孔容占比见图6,其中孔容占比中孔隙直径的划分参考文献[15]。累积孔体积反映了总孔体积与孔径的关系,而微分孔体积反映了各个区段孔径分布及其相对孔体积。根据之前的研究[6],UDP在400~1 300 nm范围是纹孔室尖端位置与纹孔口的孔径分布,在微分孔体积中可以明显看出,LDP、HDP在此范围原有孔径的基础上形成了更多孔体积,尤其是HDP较为明显,推测是由于脱木质素对于纹孔结构和木纤维的破坏导致的。此外,UDP在1 300~4 000 nm几乎没有孔径分布,LDP和HDP有明显的孔径分布和体积变化,特别是LDP和HDP在2 400~17 000 nm,相比低强度脱木质素的LDP,高强度脱木素的HDP孔体积变化更显著,这与在荧光显微镜中所观察到的细胞角隅和胞间层由于木质素的脱出而产生的孔隙一致。同时,在孔容占比图中也看出脱木质素明显增大了1 000~21 000 nm范围的孔隙,进而提高了孔隙率。
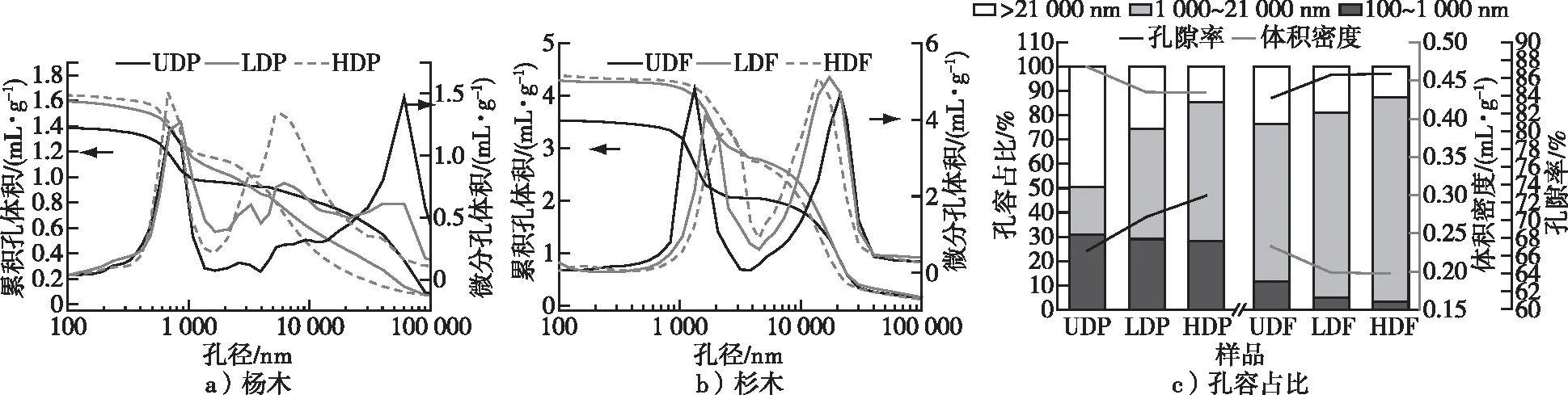
图6 杨木、杉木孔径与累积孔体积、微分孔体积的关系和孔容占比Fig. 6 The relationship between pore size and cumulative pore volume, differential pore volume, and pore volume proportion of poplar and Chinese fir
从图6中还可见,脱木质素处理对于杉木材累积孔体积与微分孔体积也有较大影响。杉木UDF在100~3 000 nm的孔隙应为纹孔膜上小孔与纹孔口的孔径分布,4 000~40 000 nm为晚材和早材间的孔隙[1]。相比HDF,UDF、LDF在20 000~36 000 nm微分孔体积分布曲线完全重合,表明脱部分木质素处理对于直径较大的早材孔径分布影响较小。然而,脱木质素对于纹孔膜和纹孔口有明显破坏,随着脱木质素程度提高,LDF和HDF在纹孔结构的基础上形成了更大的孔径分布范围。此外,UDF在3 000~4 000 nm处几乎没有孔隙,脱木质素之后的LDF和HDF出现了明显的孔径分布。分析原因是杉木材细胞角隅、胞间层等位置木质素脱出导致的,与荧光显微镜结果一致。但是与杨木材不同,杉木材的细胞角隅和胞间层间产生的孔隙大于杨木材,且在累积孔体积和孔容占比图中1 000~21 000 nm的变化也大于杨木,这主要是杉木材的木质素脱出量大于杨木材导致的。
2.5 吸湿性试验
脱木质素前后杨木材与杉木材的吸湿性变化见图7,可以看出,UDF和UDP在各湿度下的吸湿平衡含水率略低于相关研究,可能是由于热水煮沸和干燥导致的半纤维素及活性羟基量变化所导致的[16]。脱木质素增大了杨木材与杉木材的吸湿平衡含水率,脱木质素程度越高,吸湿平衡含水率增加越多。相比UDF,LDF各湿度下的吸湿平衡含水率增加了1.25%~6.87%,HDF的增加了3.23%~15.92%;而相比UDP,LDP吸湿平衡含水率增加了0.88%~10.10%,HDP的增加了12.25%~21.14%。脱木质素处理后速生材吸湿性的提高可能是由于木质素部分脱出后羟基可及性增高导致的[17]。同时,红外光谱也表明脱木质素后,半纤维素及纤维素的相对含量明显增加,表明以纤维素和半纤维素为主的吸湿性物质并未减少。另一方面,脱木质素处理在木质素间微孔的基础上产生大量的介孔和大孔,特别是HDF和HDP的介孔体积增量显著高于低强度脱木质素的介孔体积,而脱木质素处理材的吸湿性提升率并没有介孔的变化显著。这可能是由于非结晶区是水分存在的主要区域,在微纤丝间的间隙中存在着大量的木质素,介孔来源于微纤丝间隙,表明微纤丝间介孔体积的大量增加可能对吸湿性的作用并不明显,特别是在中低湿度下以羟基为结合的水分吸附范围。而脱木质素处理后纤维素结晶度的提高说明结晶区占比的变大,破坏了无定型区,也增加了结晶区对水分吸附的限制。
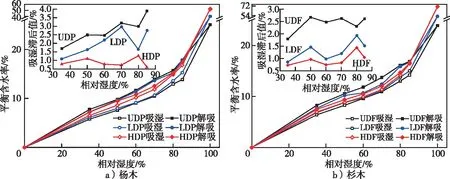
图7 未处理和脱木质素处理杨木和杉木的吸湿性Fig. 7 The hygroscopicity of untreated and delignification-treated poplar and Chinese fir
脱木质素处理对于杨木材和杉木材的吸湿性影响有较大差异。UDP的吸湿平衡含水率在相对湿度35%~85%下均小于UDF,在相对湿度35%~65%下,脱木质素处理的杉木材吸湿平衡含水率均大于杨木材,而在相对湿度70%~85%,HDP的吸湿平衡含水率大于HDF。这可能是由于杨木的半纤维素含量大于杉木,高强度脱木质素处理后,随着相对湿度的增加,杨木半纤维素膨胀后可以结合更多的水分导致的。
从图7杨木材与杉木材的吸湿性中还可以看出,脱木质素程度越高,吸湿滞后值越小。在相对湿度70%下,相比UDF的滞后值,LDF和FDF分别下降54%和68%,LDP和HDP相比UDP的滞后值分别下降7%和77%。脱木质素处理主要通过提高吸湿平衡含水率和降低解吸平衡含水率,进而降低了吸湿滞后。而相关学者[16-18]也观察到了低木质素含量的植物纤维材料吸湿滞后较小,这主要是由于脱木质素处理后,作为细胞壁中结壳物质的木质素减少使细胞壁基体的刚性降低,减少了吸湿和解吸过程中细胞壁对于水分子的蒸发和吸收响应的差异程度,进而降低了吸湿滞后。
2.6 脱木质素处理材Hailwood-Horrobin等温吸附曲线拟合分析
Hailwood-Horrobin模型不仅可以解析出吸湿过程中的单分子层吸附和多分子层吸附,还可以表征吸湿过程中羟基基团的变化,从而更有利于探究吸湿过程中的本质变化。基于Hailwood-Horrobin模型拟合计算的等温吸附曲线参数见表2,根据表2中的参数可以看出,H-H拟合出的吸湿性与35%~85%实测值相关性较高,R2均达到了99%以上,同时从杨木与杉木拟合曲线(图8)可以看出,吸湿曲线类型属于典型的“SII”型等温吸附线。
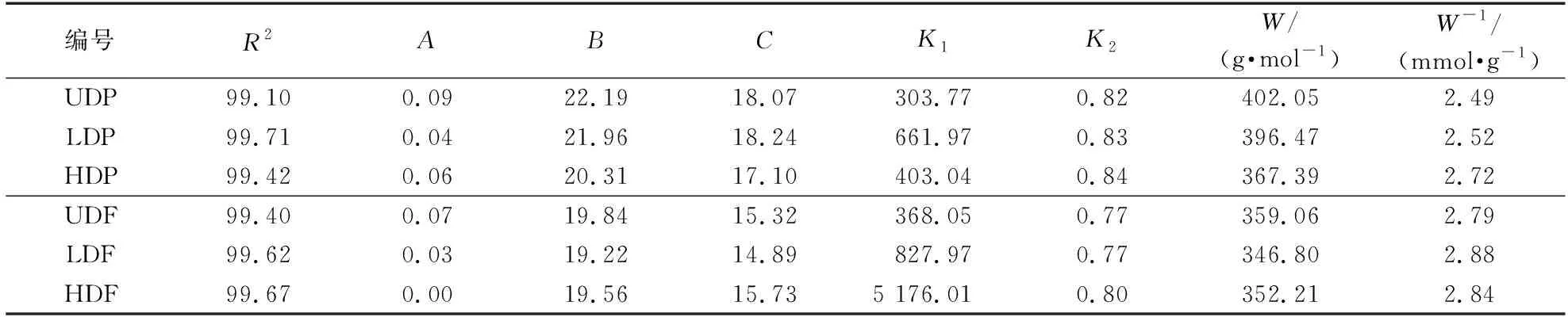
表2 基于Hailwood-Horrobin模型拟合计算的等温吸附曲线参数Table 2 Fitting calculation of isothermal adsorption curve parameters based on the Hailwood-Horrobin model
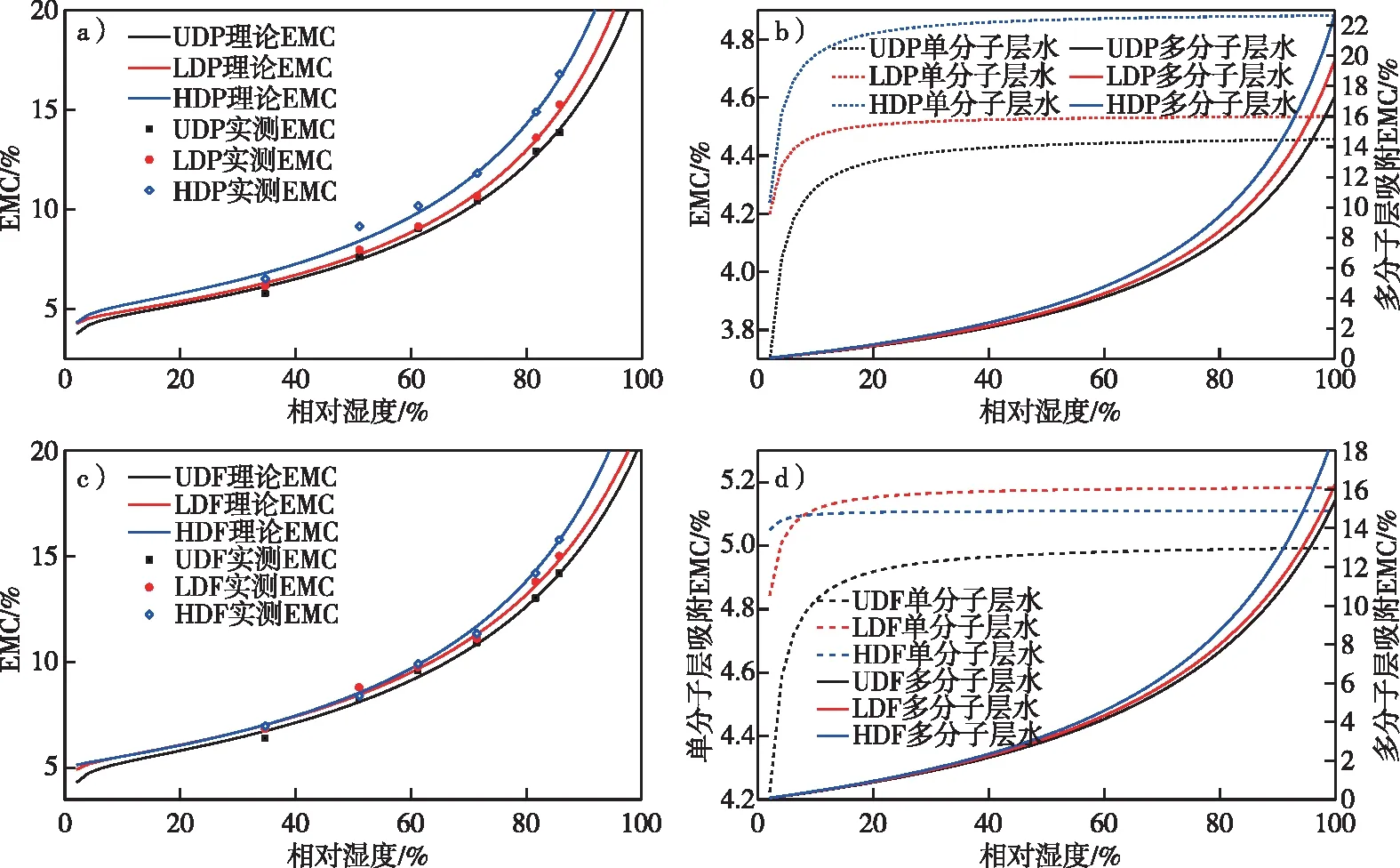
图8 脱木质素处理前后杉木材和杨木材吸湿理论拟合和单分子层与多分子层水分吸附曲线Fig. 8 The equilibrium moisture content and monomolecular and polymolecular adsorption isotherms for Chinese fir and poplar before and after delignification treatment
从图8的单、多分子层吸附水及平衡含水率拟合曲线中可以看出,脱木质素处理增强了杨木材与杉木材的吸湿性:相比UDP,LDP和HDP的单分子层水分提升了1.76%和9.42%,多分子层水分提升了14.36%和33.23%;而相较于UDF,LDF、HDF的单分子层水分提升了3%左右,多分子层水分提升了5.72% 和21.13%。这表明脱木质素处理后多分子层水分吸附能力的变化量大于单分子层,同时,脱木质素处理后杨木的多分子层水吸附能力强于杉木,与在相对湿度70%~85% HDP的吸湿平衡含水率大于HDF的一致。这可能是由于脱木质素后细胞壁的硬度降低,随着相对湿度的增加,更有利于半纤维素的膨胀,而杨木的半纤维素含量大于杉木,造成了杨木材和杉木材多分子层吸附的差异。此外,相较于未处理材,HDP单分子层吸附水的变化量大于LDP,而LDF单分子层吸附水的变化量大于HDF,与代表羟基可及性的1/W和吸湿性较强的半纤维素在红外光谱中的相对含量变化趋势一致。
由于针叶材和阔叶材木质素含量的差异,导致了脱木质素材在细胞角隅和胞间层等位置以及细胞壁中介孔等位置的微观孔隙差异;同时,由于杨木和杉木半纤维素含量的差异导致脱木质素材吸湿性中单分子层和多分子层的差异,但是脱木质素并没有改变杨木结晶度大于杉木的本质。
3 结 论
1)脱木质素处理提高了杨木材和杉木材的吸湿平衡含水率,同时显著降低了吸湿滞后。在相对湿度35%~85%时,UDP吸湿平衡含水率小于UDF;在相对湿度35%~60%时,LDF和HDF的吸湿平衡含水率大于LDP和HDP;但是在相对湿度70%~85%时,由于脱木质素处理的杨木材多分子层吸附能力的提升,导致HDP的吸湿平衡含水率大于HDF。同时,H-H模型证明了脱木质素对于杨木材和杉木材多分子层吸附能力的提升大于单分子层,脱木质素处理对杨木材多分子层水的吸附能力提升强于杉木材。
2)脱木质素处理显著增加了2~30 nm介孔和1 000~21 000 nm大孔的数量,增加的孔隙主要来自微纤丝间隙和细胞角隅及胞间层等位置,但是脱木质素材的吸湿性远没有细胞壁中介孔的变化明显。杉木材在介孔和微孔区间的孔径变化范围和孔容增量均大于杨木材,同时,脱木质素改变了木质素在次生壁、胞间层、细胞角隅的浓度分布规律,提高了杨木材和杉木材的结晶度。红外光谱分析表明,脱木质素处理后杨木材和杉木材中半纤维素和纤维素等主要吸湿物质相对含量增大。
