水稻CRISPRa系统的优化及验证
2023-10-17姚毅菲刘林丽苏颜琦刘华伟
杨 柳,姚毅菲,刘林丽,苏颜琦,刘华伟
(西北农林科技大学 生命科学学院,陕西杨凌 712100)
通常采用组成型启动子超表达基因研究其功能[1],但有些基因难以克隆,如编码序列15 kb的拟南芥BIG基因[2]。此外,改善作物品质通常需要同时上调多个基因表达,如在番茄中同时过表达SlWRKY35和SlLCYE基因相较于单独表达单个基因,可以显著提高果实中的叶黄素含量[3]。但同时表达多个内源基因时,需要克隆多个基因、启动子和终止子,增加载体构建难度。
ZFP-TADs和TALE-TF能够激活内源基因,但均设计复杂、成本高且难进行多基因激活。近年来,基于CRISPR衍生出CRISPRa等多种技术[4-5],其中CRISPRa[6-9]相较于ZFP-TADs和TALE-TF,只需合成多个sgRNA,可快速、低成本地同时上调多个内源基因表达。
水稻作为中国主要农作物之一,对于保障粮食安全具有重要意义,而提高其产量、品质的关键在于育种技术创新[10-11]。CRISPRa与CRISPR相比,不造成DNA双链断裂,安全性更高;与转基因相比,无需引入外源基因即可上调多个基因表达。目前已有植物的CRISPRa系统,如CRISPR Act2.0、CRISPR Act3.0、dCas9-TV及CCTV等[6-9]。本研究对dCas9进行点突变减少其与DNA的非特异性结合、增强其对标准PAM(NGG)的识别,并对基因序列进行水稻密码子优化,旨在开发特异性更强、激活效率更高的水稻CRISPRa系统,为水稻及其他作物分子设计育种提供新的技术方案。
1 材料与方法
1.1 材 料
1.1.1 菌株与质粒 大肠杆菌DH5α感受态细胞购自北京博迈德基因技术有限公司;质粒pYLgRNA-OsU6a由华南农业大学刘耀光院士惠赠;质粒HBT-dCas9-CCTV-2由中山大学李剑峰教授惠赠;质粒pGL3-Basic由西北农林科技大学赵天永教授惠赠;质粒p35S-eGFP、pGR107、pUC19-dCas9、pUC19-TV、pUC19为实验室保存。
1.1.2 材料与试剂 植物材料为日本晴(OryzasativaL. japonica cv. nipponbare,NPB);限制性内切酶(BamHⅠ、EcoRⅠ、NotⅠ、SalⅠ)、RNAiso Plus、TB Green○RPremix ExTaqTMⅡ购自Takara公司;限制性内切酶BsaⅠ、T4DNA连接酶购自NEB公司;高保真聚合酶2×HiProof 2G PCR Master Mix、无缝克隆酶2×SmartSeamless Cloning Mix购自陕西杨凌杰一生物技术有限公司;反转录试剂盒UEIris RT mix with DNase购自苏州宇恒生物;CellulaseR-10、MacerozymeR-10购自Yakult公司;PEG4000购自Biotopped公司。原生质体制备溶液配方参照文献[11]。
1.1.3 引物合成及测序 引物合成及测序委托北京擎科生物科技有限公司西安分部完成。
1.2 方 法
1.2.1 点突变dCas9为HFdCas9 利用表1引物对dCas9中N497A、R661A、Q926A、D1135E进行四重点突变得到HFdCas9。以pUC19-dCas9质粒为模板进行扩增,PCR体系如下:2×HiProof 2G PCR Master Mix 25 μL,引物各 1 μL,pUC19-dCas9质粒1 μL,加ddH2O至50 μL。反应程序:95 ℃预变性5 min;95 ℃变性30 s,60 ℃退火25 s,72 ℃延伸2 min 30 s,32个循环;72 ℃延伸5 min。将扩增产物进行琼脂糖凝胶电泳,对目标条带进行胶回收。

表1 点突变dCas9为HFdCas9 所用引物Table 1 Primers used for HFdCas9 as point mutation dCas9
利用无缝克隆的方式构建载体,体系如下: 2×Smart Seamless Cloning Mix 5 μL,N497A片段0.5 μL,R661A片段0.5 μL,Q926A片段0.5 μL,D1135E片段0.5 μL,加ddH2O至10 μL。55 ℃连接30 min,65 ℃失活10 min,连接产物转化至大肠杆菌DH5α感受态细胞,涂至氨苄抗性平板。37 ℃过夜培养后挑取单克隆摇菌,提取完质粒并进行测序验证。
1.2.2 水稻原生质体HFdCas9-TV表达载体构建 利用表2引物构建原生质体HFdCas9表达载体。从pGL3-Basic质粒扩增2×35S片段,从上文所构建的pUC19-HFdCas9质粒扩增HFdCas9片段,从p35S-eGFP质粒扩增NOS-Ori-Amp片段,用无缝克隆的方式构建HFdCas9表达载体。PCR体系和程序同“1.2.1”。无缝克隆体系:2×SmartSeamless Cloning Mix 5 μL, 2×35S片段0.5 μL,HFdCas9片段2 μL,NOS-Ori-Amp片段1 μL,加ddH2O至10 μL。无缝克隆程序及转化验证同“1.2.1”。

表2 HFdCas9表达载体构建所用引物Table 2 Primers for construction of HFdCas9 expression plasmid
利用表3引物构建原生质体HFdCas9-TV表达载体,在dCas9序列后插入TV结构域和HA标签。从pUC19-TV质粒扩增TV片段,从pGR107质粒扩增2×HA片段,将构建的2×35S-HFdCas9质粒用BamHⅠ单酶切。进行琼脂糖凝胶电泳,胶回收TV片段、HA片段、线性化的2×35S-HFdCas9片段。利用无缝克隆的方式构建载体,体系如下:2×SmartSeamless Cloning Mix 5 μL,线性化的2×35S-HFdCas9 2 μL,TV片段1 μL,HA片段1 μL,加ddH2O至10 μL。无缝克隆程序及转化验证同“1.2.1”。
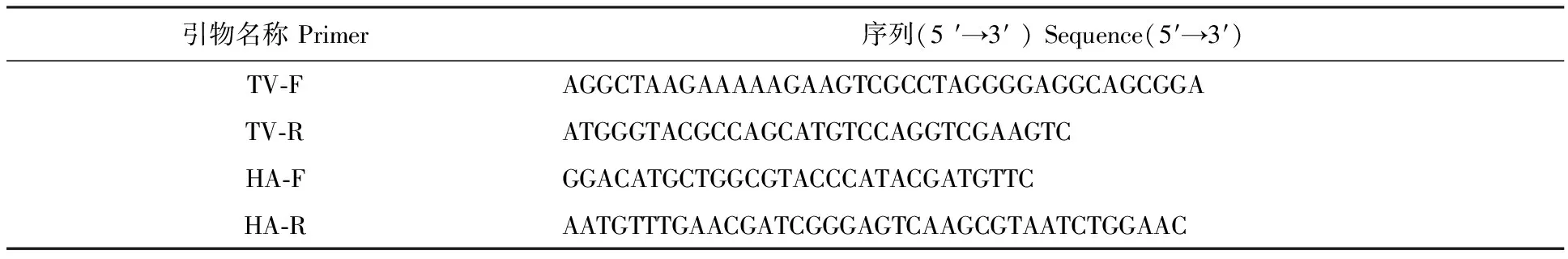
表3 HFdCas9-TV表达载体构建所用引物Table 3 Primers for construction of HFdCas9-TV expression plasmid
1.2.3 pUC19-sgRNA载体构建 将表4中OsGW7sgRNA-F/R干粉引物浓度稀释至100 μmol/L,吸取ddH2O 8 μL,OsGW7sgRNA-F/R引物各1 μL。95 ℃加热2 min,缓慢降温至25 ℃。吸取1 μL的sgRNA引物退火产物,1 μL的pYLgRNA-OsU6a质粒,0.5 μL的BsaⅠ-HF限制性内切酶,1 μL的T4DNA Ligase,1 μL的 10×T4DNA Ligase Buffer,1 μL的10×CutSmart Buffer,加ddH2O至10 μL,进行金门组装。金门组装程序:37 ℃酶切5 min,20 ℃连接5 min,10个循环。将上述产物作为PCR模板,以U6a-F/R引物进行扩增。PCR体系:2×HiProof 2G PCR Master Mix 25 μL,模板1 μL,引物U6a-F/R各1 μL,加ddH2O至50 μL。反应程序:95 ℃预变性5 min;95 ℃变性25 s,51 ℃退火25 s,72 ℃延伸20 s,共35个循环;72 ℃延伸5 min。进行琼脂糖凝胶电泳与胶回收。以pUC19质粒为载体,U6a-F/R扩增产物为片段,均用EcoRⅠ和SalⅠ进行酶切和胶回收后,于22 ℃连接2 h,转化验证同“1.2.1”。OsF3HsgRNA载体构建方法同OsGW7。
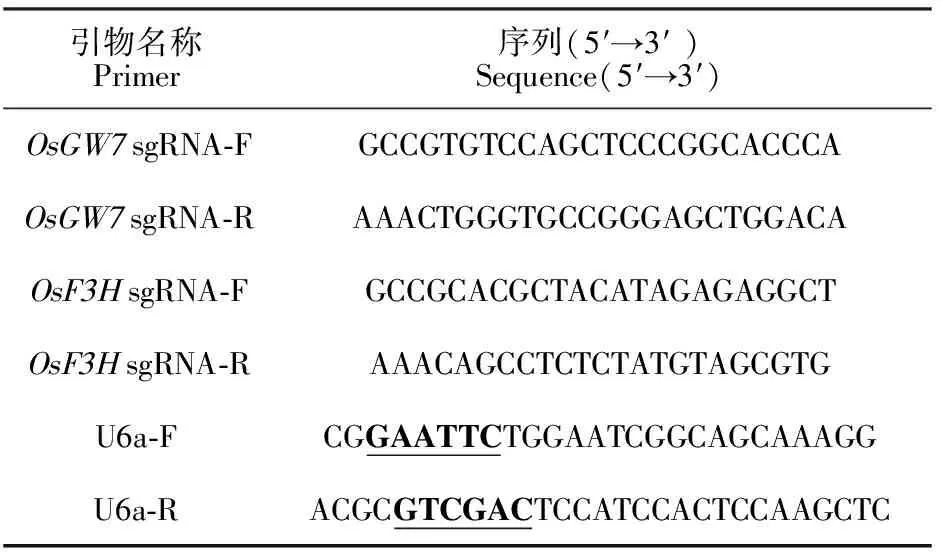
表4 pUC19-sgRNA载体构建所用引物Table 4 Primers for construction of pUC19-sgRNA plasmid
1.2.4 水稻原生质体的制备与转染 参照孙鹤等[12]的方法进行部分改动。取培养10 d的水稻幼苗,剪去根部和叶片,保留叶鞘部分,用水冲洗干净。用滤纸吸干水分,用刀片将叶鞘切成 0.5 mm的小段,转移至有50 mL 0.6 mol/L甘露醇溶液的锥形瓶中,置于摇床中25 ℃、60 r/min缓慢摇动30 min。用200目的尼龙膜过滤除去锥形瓶中甘露醇溶液,加入15 mL酶解液,在摇床中25 ℃、60 r/min缓慢摇动4 h。
吸取10 μL酶解液至载玻片,用光学显微镜观察酶解程度和原生质体状态。酶解充分后,加入等体积预冷的W5溶液,置于摇床,加大转速至80 r/min摇动5 min。用双层尼龙膜过滤残渣,收集原生质体至50 mL离心管中。将离心机设置为加速度a=3 200 g离心7 min,收集原生质体。弃上清,重新加入10 mL W5溶液,将原生质体悬浮,显微镜下检验。200 g离心7 min,收集原生质体,再次弃上清,加入10 mL W5溶液,悬浮原生质体,放置于冰上,避光静置40 min。 200 g离心7 min,收集原生质体,弃上清。加入2 mL MMG溶液,取10 μL溶液用血细胞计数板计数,将原生质体浓度稀释到2×106个/mL,即可用于后续原生质体转染。
HFdCas9-TV质粒和sgRNA质粒各取 16 μg至2 mL离心管底,加入200 μL原生质体,轻轻混匀。加入220 μL PEG4000转染溶液,将其混匀。黑暗环境下,室温静置25 min。加入 800 μL W5溶液,缓慢颠倒混匀。将离心机加速模式设置为soft模式,200×g离心5 min,收集原生质体。加入1 mL W1溶液,轻轻混匀,黑暗环境下室温静置12 h。
1.2.5 水稻原生质体RNA的提取及RT-qPCR 将过夜培养的原生质体200×g离心10 min收集于管底,弃上清。加入1 mL RNAiso Plus,转移至RNase-free的1.5 mL离心管中,冰上静置10 min。加入500 μL的氯仿,用力晃动至呈现乳白色,冰上静置10 min。4 ℃,12 000 r/min离心15 min,取出离心管,管内液体分为三层,吸取上清至另一离心管中。加入等体积的异丙醇,颠倒混匀后,-20 ℃冰箱沉淀4 h。4 ℃,12 000 r/min离心15 min,弃上清,管底可见白色沉淀。加入RNase-free水配置的75%乙醇,洗涤沉淀, 4 ℃,12 000 r/min离心10 min,弃上清。重复上步操作一次,放置于超净台中,吹干乙醇。加入20 μL的RNase-free水溶解沉淀,用Nandrop测定RNA浓度,取部分样品进行琼脂糖电泳检测其质量。
参照反转录试剂盒UEIris RT mix with DNase说明书进行反转录,参照TB Green○RPremix ExTaqTMⅡ说明书进行qRT-PCR,引物见表5。OsUBQ作为内参基因,利用2-ΔΔCt法计算目的基因表达量变化。
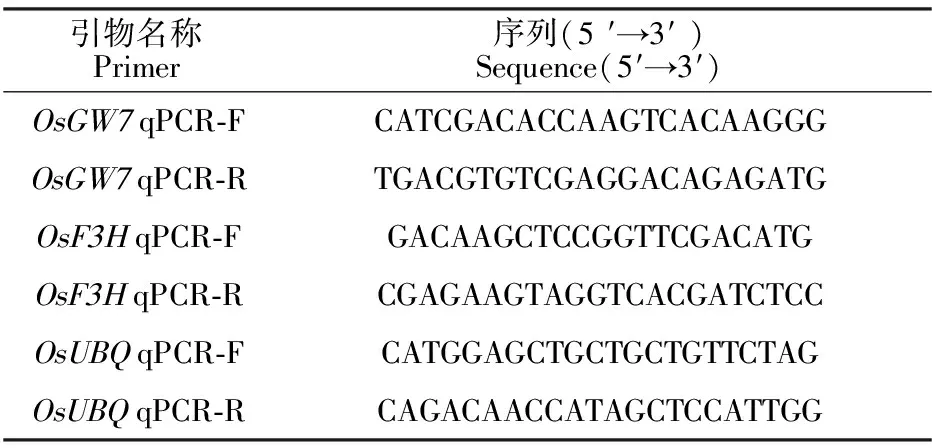
表5 qRT-PCR所用引物Table 5 Primers used for qRT-PCR
2 结果与分析
2.1 CRISPRa载体构建
2.1.1 HFdCas9-TV表达载体构建 通过引物引入dCas9的N497A、R661A、Q926A、D1135E 4个氨基酸位点突变,经测序验证,成功获得连接在pUC19上的4个点突变的HFdCas9(图1-a)。经PCR扩增得到启动子2×35S片段、HFdCas9片段以及NOS-Amp-Ori片段,大小分别为909 bp、4 223 bp、1 973 bp(图1-b)。通过无缝克隆构建 2×35S-HFdCas9载体,经测序验证,载体构建成功。

a:dCas9中N497A、R661A、Q926A、D1135E 4个位点突变后的测序峰图;b:M.Marker;1.HFdCas9;2.NOS-Amp-Ori; 3.2×35S;c:M.Marker;1.TV;2.2×HA
将上步所得质粒用BamHⅠ单酶切,扩增TV结构域和2×HA标签片段。因TV结构域含有较多重复序列,扩增产物出现多个条带,取大小正确的1 404 bp条带用于载体构建;2×HA片段大小为93 bp(图1-c)。通过无缝克隆构建2×35S-HFdCas9-TV载体,经测序验证,载体构建成功。
2.1.2 pUC19-sgRNA载体的构建 HFdCas9-TV表达载体大小为8 588 bp,质粒过大会影响转染效率。因此将转录sgRNA的质粒和表达dCas9-TV的质粒共转原生质体(图2-a)。利用金门组装将sgRNA连接至pYLsgRNAU6a载体,反向PCR获得顺序正确的sgRNA表达盒,连接至pUC19载体。sgRNA表达盒的片段大小约为608 bp(图2-b)。
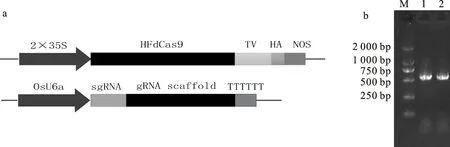
a:HFdCas9-TV表达载体和pUC19-sgRNA载体示意图
2.2 CRISPRa激活能力的验证
原生质体制备完成后,转染前需在光学显微镜下检验(图3-a)。经检验,本研究所制备的水稻原生质体细胞数量充足,形态完整,可用于后续CRISPRa质粒转染。使用Nandrop测定提取的RNA样品A260/280比值,均在1.9左右。进一步通过琼脂糖凝胶电泳检测RNA质量(图3-b),结果显示其RNA质量合格。

a:原生质体镜检图(10倍物镜、10倍目镜的光学显微镜)
利用所构建HFdCas9-TV靶向激活OsGW7,通过RT-qPCR分析,发现基因OsGW7表达量上调至对照组的95倍;利用CCTV系统在相同情况下,OsGW7表达量可上调至对照组43倍,激活效率提升至CCTV的2.2倍(图4-a)。进一步利用HFdCas9-TV靶向激活OsF3H,OsF3H表达量上调至对照组的46倍(图4-b)。经OsGW7和OsF3H激活验证,证明所构建CRISPRa系统具有激活能力。将OsGW7、OsF3HsgRNA质粒同时与HFdCas9-TV质粒共转至水稻原生质体,OsGW7、OsF3H分别被激活至对照组的33倍、26倍(图4-c),证明所构建CRISPRa系统具有多重激活能力。

a. OsGW7的转录激活;b. OsF3H的转录激活;c. OsGW7和 OsF3H同时转录激活;* *代表在P=0.01水平有显著差异
3 讨 论
高效sgRNA的设计和筛选是CRISPRa成功激活目的基因的关键,其靶点通常选择在转录起始位点前250 bp内[6,8]。OsGW7的靶点[6]位于转录起始位点前的[-73,-93]位置,OsF3H的靶点[8]与常规的设计不同,其位于[+91,+72](https://www.ncbi.nlm.nih.gov/genome/gdv/browser/gene/?id=9270463),在转录起始位点后的5′UTR,仍具有激活能力。后续可研究5′UTR位置靶点是否普遍具有激活能力,进一步拓展靶点设计范围。
本研究构建由HFdCas9-TV、pUC19-sgRNA组成的水稻CRISPRa系统。前者大小为8.3 kb,其中HFdCas9-TV片段为5.6 kb。为了优化其表达采用2×35S强启动子且进行水稻密码子优化。采用文献[6,8]中OsGW7和OsF3H的sgRNA,在水稻原生质体水平对所构建CRISPRa系统进行功能验证,两个基因表达均被成功上调。相较于CCTV系统[9],对OsGW7激活效率提高至其2.2倍,从43倍提升至95倍。相较于CRISPR-Act 3.0系统[8]激活OsF3H至对照组120倍,本研究构建的CRISPRa系统激活至对照组46倍。分析原因可能是CRISPR-Act3.0利用Suntag招募到更多转录激活域,覆盖更广的启动子区域。后续将对本研究构建CRISPRa系统进一步优化并提高其效率。
目前已将基因编辑技术应用于水稻、小麦等作物改良[6,13]。运用CRISPR技术敲除感病基因“做减法”虽可使作物抗病但出现了早衰、产量下降等负面表型[14],CRISPRa技术可快捷地对作物“做多重加法”,通过同时激活抗逆、抗病、产量相关基因获得聚合性状的优良作物。此外,对于作物育种,控制目标基因的转录强度至关重要。如关键抗病基因表达需要适当水平上调,过度转录会引发细胞的程序性死亡甚至影响植物的正常发育[15-16]。CRISPRa技术通过在基因启动子区招募RNA聚合酶以提高其转录水平的表达,相较于超表达更接近生理水平,调控更自然,避免能量浪费及代谢损害。因此,调控自然的“做多重加法”更适合于作物性状改良。
