Genome-wide association study for numbers of vertebrae in Dezhou donkey population reveals new candidate genes
2023-10-16SUNYanLlYuhuaZHAOChanghengTENGJunWANGYonghuiWANGTianqiSHlXiaoyuanLlUZiwenLlHaijingWANGJijingWANGWenwenNlNGChaoWANGChangfaZHANGQin
SUN Yan ,Ll Yu-hua ,ZHAO Chang-heng ,TENG Jun ,WANG Yong-hui ,WANG Tian-qi ,SHl Xiaoyuan,LlU Zi-wen,Ll Hai-jing,WANG Ji-jing,WANG Wen-wen,NlNG Chao,WANG Chang-fa#,ZHANG Qin#
1 Shandong Provincial Key Laboratory of Animal Biotechnology and Disease Control and Prevention,College of Animal Science and Technology,Shandong Agricultural University,Tai’an 271018,P.R.China
2 Liaocheng Research Institute of Donkey High-Efficiency Breeding and Ecological Feeding,Liaocheng University,Liaocheng 252059,P.R.China
3 National Engineering Research Center for Gelatin-Based Traditional Chinese Medicine,Dong-E E-Jiao Co.Ltd.,Liaocheng 252200,P.R.China
4 Shandong Provincial Key Laboratory of Animal Disease Control and Breeding,Institute of Animal Science and Veterinary Medicine,Shandong Academy of Agricultural Sciences,Jinan 250100,P.R.China
Abstract Numbers of vertebrae is an important economic trait associated with body size and meat productivity in animals. However,the genetic basis of vertebrae number in donkey remains to be well understood. The aim of this study was to identify candidate genes affecting the number of thoracic (TVn) and the number of lumbar vertebrae (LVn) in Dezhou donkey. A genome-wide association study was conducted using whole genome sequence data imputed from low-coverage genome sequencing. For TVn,we identified 38 genome-wide significant and 64 suggestive SNPs,which relate to 7 genes (NLGN1,DCC,SLC26A7,TOX,WNT7A,LOC123286078,and LOC123280142). For LVn,we identified 9 genome-wide significant and 38 suggestive SNPs,which relate to 8 genes (GABBR2,FBXO4,LOC123277146,LOC123277359,BMP7,B3GAT1,EML2,and LRP5). The genes involve in the Wnt and TGF-β signaling pathways and may play an important role in embryonic development or bone formation and could be good candidate genes for TVn and LVn.
Keywords: numbers of vertebrae,GWAS,genotype imputation,Dezhou donkey
1.Introduction
During livestock evolution,the body size of domestic animals varied greatly between and within species(breeds). The number of vertebrae,especially the number of thoracolumbar vertebrae,greatly affects the carcass length (Borcherset al.2004). Variations in number of thoracolumbar vertebrae have been reported in pigs (King and Roberts 1960),sheep (Donaldsonet al.2013) and cattle (Yanget al.2015). Different numbers of vertebrae have also correlations with growth and meat production(Borcherset al.2004) and therefore variation in number of thoracolumbar has been considered as a selection trait in commercial pig breeding (Hiroseet al.2013;Burgoset al.2015). In the genusEquus,the common number of thoracic vertebrae (TVn) is 18,while 17 and 19 were also reported (Stecher 1962). In domestic horse,zebras and hybrids,animals with 6 number of lumbar vertebrae(LVn) were more observed than those with 5 LVn (Sisson and Grossman 1953),while in donkey and hemionus,the common LVn is 5 (Jamdar and Ema 1982).
For thousands of years,donkeys had been used for transporting goods and facilitating communications in distant areas. China has a history of raising donkeys for 4 000 years and has abundant local donkey breeds.Dezhou donkey,originated in Shandong Province,is one of the most famous donkey breeds in China. It is characterized by its large body size,good adaptability,and resistance of roughage. It has been introduced into other places of China as superior breeding stocks for genetic improvement of other local donkey breeds. Nowadays,donkey industry is playing more and more important roles in animal husbandry in China due to donkey’s multiple valuable products (skin,milk,and meat). As a result,selective breeding is gradually becoming an important issue in donkey production and some breeding work has been carrying out in Dezhou donkey population in recent years.
Previous studies showed that in the Dezhou donkey population there also exist variations in TVn and LVn.From a survey of 455 donkeys (Liuet al.2022),it was found that there were 5 types of the combination of TVn and LVn,i.e.,T18L5 (18 thoracic vertebrae and 5 lumbar vertebrae,75.8%),T18L6 (8.8%),T17L6 (11.9%),T17L5(2.4%),and T19L5 (1.1%). It was also observed that the donkey’s body length and carcass weight were influenced by vertebral number (Liuet al.2022). These evidences suggest that selection for the number of thoracolumbar vertebrae may benefit the donkey industry by increasing body size,which induces researches on the genetic mechanism of number of thoracolumbar vertebrae in donkey.
Genome-wide association study (GWAS) based on high density of SNP markers has been one of the most commonly used strategies for identifying genes for complex traits (Changet al.2018). Typically,the SNP genotypes are obtained by using high (or medium)density SNP chips and many commercial SNP chips have been developed for major farm animal species(Stock and Reents 2013). However,there are still some species,such as donkey,for which no such array is available,which inhibits the application of GWAS in these species. Moreover,the limited number of SNPs of a chip do not cover all variants and genomic regions associated with the traits,which will reduce the power to identify traits-related variants and regions. Recently,along with the rapid development of next generation sequencing technology and reduction of sequencing cost,GWAS using genotypes revealed by whole genome sequencing (WGS),instead of SNP array,has drawn interests of animal GWAS community. It has been shown that using WGS data would improve the power of GWAS and increase the ability for mapping causal mutations(Daetwyleret al.2014). However,high-coverage WGS is still too expensive for large scale genomic analysis,especially for livestock (Fraseret al.2018). An alternative is to perform low coverage genome sequencing (lcWGS)at about 1× or less,and then recovering the missing genotypes by imputation. This approach has been used in human and some animal species and proven to be a feasible alternative to normal sequencing (Nicodet al.2016;Liuet al.2018;Zhanget al.2021). Therefore,the objective of this study was to identify genomic regions and candidate genes associated with the variation of thoracic and lumbar vertebrae numbers using imputed sequencing data in Dezhou donkeys. The identified candidate genes and variants were expected to lay a foundation for the molecular breeding for thoracic and lumbar vertebrae numbers in donkeys.
2.Materials and methods
2.1.Population and phenotypic data collection
Animals (Dezhou donkeys) used in this study were obtained from the Black Donkey Research Institute,Dong-E E-Jiao Co.,Ltd.(Liaocheng,Shandong,China).The dataset of numbers of thoracolumbar vertebrae consisted of two parts. One contained data of 202 male donkeys from several different fattening herds. These animals were slaughtered in the same abattoir,their skeletons were isolated and thoracolumbar vertebrae were counted. The other part contained the data of 145 donkeys from a breeding herd. These animals were scannedinvivousing Digital Radiography (DR,x-ray) and their thoracolumbar vertebrae were counted on the scan pictures. For all animals,blood samples were collected from jugular vein using anticoagulation tubes. All blood samples were stored at–20°C for the extraction of total DNA.
2.2.Sequencing and mapping
The extracted DNA samples were sheared,end repaired,A-tailed and ligated to Illumina paired end adapters(Illumina,CA,USA) and then constructed 350-bp DNA library. Whole genome sequencing was performed on an Illumina NovaSeq 6000 platform (Illumina) following a standard protocol. The length of the sequence reads was 150 bp. Clean data were obtained after removing poor quality reads,adaptors,and more N bases reads using Trimmomatic (v0.39) (Bolgeret al.2014). Read quality was assessed through FastQC Program (v0.11.9).Subsequently,clean reads were mapped to donkey reference genome (Wanget al.2020) using BWA (v0.7.17)(Li and Durbin 2009). QualiMap (v.2.2.1,http://qualimap.conesalab.org/) (Okonechnikovet al.2016) was used to evaluate sequencing alignment data. BAM files were sorted with SAMTools (v1.11) (Liet al.2009) and marked and the duplicates were removed using Picard (v2.26.10,https://github.com/broadinstitute/picard/releases/tag/2.26.10). These bam files were used for subsequent genotype imputation.
2.3.Genotype imputation and evaluation of imputation accuracy
High-quality reads were used for SNP calling with BaseVar (v0.8.0) (https://www.cnpython.com/pypi/basevar). We used STITCH (v1.6.3) (Davieset al.2016)to impute the missing genotypes assuming 10 ancestral haplotypes. Since there were still a high proportion of missing genotypes after this imputation,we performed second imputation using Beagle (v5.1) (Browning and Browning 2016) following the suggestion of Tengetal.(2022). The resulted SNP genotype data were qualitycontrolled by removing SNPs with minor allele frequency(MAF)<0.05,imputation info_score<0.4,and a Hardy-Weinberg Equilibrium (HWE)P-value<1e–6.
We evaluated the imputation accuracy using sequence data of additional 18 donkeys,which were sequenced at high depth of 11.2×–16.3× (13.5× on average). We randomly removed their genotypes of some SNPs such that their sequencing depths were equivalent to that of the 347 individuals mentioned above,and then we imputed the missing genotypes of these 18 individuals along with the GWAS individuals. After imputation,we compared the imputed genotypes of the 18 individuals with their true genotypes to evaluate the imputation accuracy.The imputation accuracy was measured with genotypic correlation which is defined as squared Pearson correlation coefficient (r2) between expected dosages(posterior expectation of the imputed allele dosages)and typed genotypes (Browning and Browning 2009).Chromosomes 1,19 and 30,which represented the long,short and medium chromosomes among the donkey chromosomes,respectively,were chosen to evaluate the imputation accuracy.
2.4.Population stratification analysis
Since the animals involved in this study were from different herd with unknown genetic background,in particular,most of the data was from the abattoir and no pedigree was available for these animals,there might exist population stratification among these animals. To avoid the possible influence of population stratification on GWAS results,we used principle component analysis(PCA) to explore the potential population stratification using the SNP genotype data (Priceet al.2006). The population stratification would be accounted for by using the first two PCs as covariables in the GWAS models as suggested by Bani-Fatemietal.(2016) (see below).
2.5.GWAS analysis
Two models were used for the GWAS analysis. The first model is a regular linear mixed model. By using this model,the categorical phenotypes were treated as continuous traits. The model for a given SNP is:
whereyis the vector of phenotypic values of all animals;μis the overall mean;1is a vector with all values of 1,p1andp2are vectors of the first and second PC values of all animals,respectively;b1andb2are corresponding regression coefficients;gis the allele substitution effect of the given SNP;xis the vector of genotypes (coded as 0,1,and 2 for the three types of genotypes) of all animals for the SNP;ais the vector of additive polygenetic effects,which was assumed to be distributed asa~N(0,Gσa2)withGbeing the genomic relationship matrix that was constructed based on all imputed SNPs using the method of VanRaden (2008) andσa2being the additive genetic variance;eis the vector of random residual effects with distribution of N(0,lσe2) withlbeing an identity matrix andσe2the residual variance. The GEMMA Software(v0.98.1,https://stephenslab.uchicago.edu/software.html)(Zhou and Stephens 2012) was employed for the GWAS analysis using this model.
The second model is a logistic mixed model which is commonly used for the analysis of binary (type 1 and type 2) phenotypes. The model is as follows:
whereπis the probability that an animal having type 1 phenotype given its corresponding first and second PC values (p1andp2),SNP genotype (g) and polygenetic effect (a). The GMMAT Software (https://github.com/hanchenphd/GMMAT;Chenet al.2016) was employed for this analysis.
The analysis of LVn was conducted using animals with 18 thoracic vertebrae,including 183 L5 and 67 L6 individuals. The analysis of TVn was conducted using animals with 5 LVn,including 183 T18 and 17 T17 individuals. The Bonferroni correction (Hayes 2013)was used to determine the significance threshold and quantile–quantile (Q–Q) plots were built to check potential false positives due to population stratification.
2.6.Candidate gene identification
Genes within the 500 kb windows covering upstream and downstream of significant SNPs were identified based on the donkey genome assembly (Wanget al.2020) using SnpEff Software (4.3t (build 2017-11-24)) (Cingolaniet al.2012).
3.Results
3.1.Population phenotypic data
Numbers of thoracolumbar vertebrae of the 347 Dezhou donkeys are presented in Table 1. Again,we observed 5 types of the thoracolumbar vertebrae combinations,i.e.,T18L5,T18L6,T17L5,T17L6,and T19L5 in the study population.
3.2.The sequencing data
After sequencing data quality control,23.55 Gb clean reads were obtained from all samples. The average Q30 of each sample reached 91.30%. The average sequencing depth of all samples was 4×,ranging from 2× to 7×. Almost all reads (99.64%) were successfully mapped to the donkey reference genome.
3.3.Accuracies of genotype imputation
We obtained 9 478 127 variants after imputation usingSTITCH and Beagle. The imputation accuracies for Chr1,19,and 30 are shown in Fig.1 and the averager2were 0.978,0.977,and 0.978,respectively. After quality control,6 220 779 SNPs were retained. The distribution of these SNPs on each chromosome are shown in Fig.2.We further estimated the effective number of SNPs by applying LD (linkage disequilibrium) pruning withusing PLINK (v1.90) (Changet al.2015). 353 672 SNPs were remained,which were used for determining the significance threshold by Bonferroni correction.
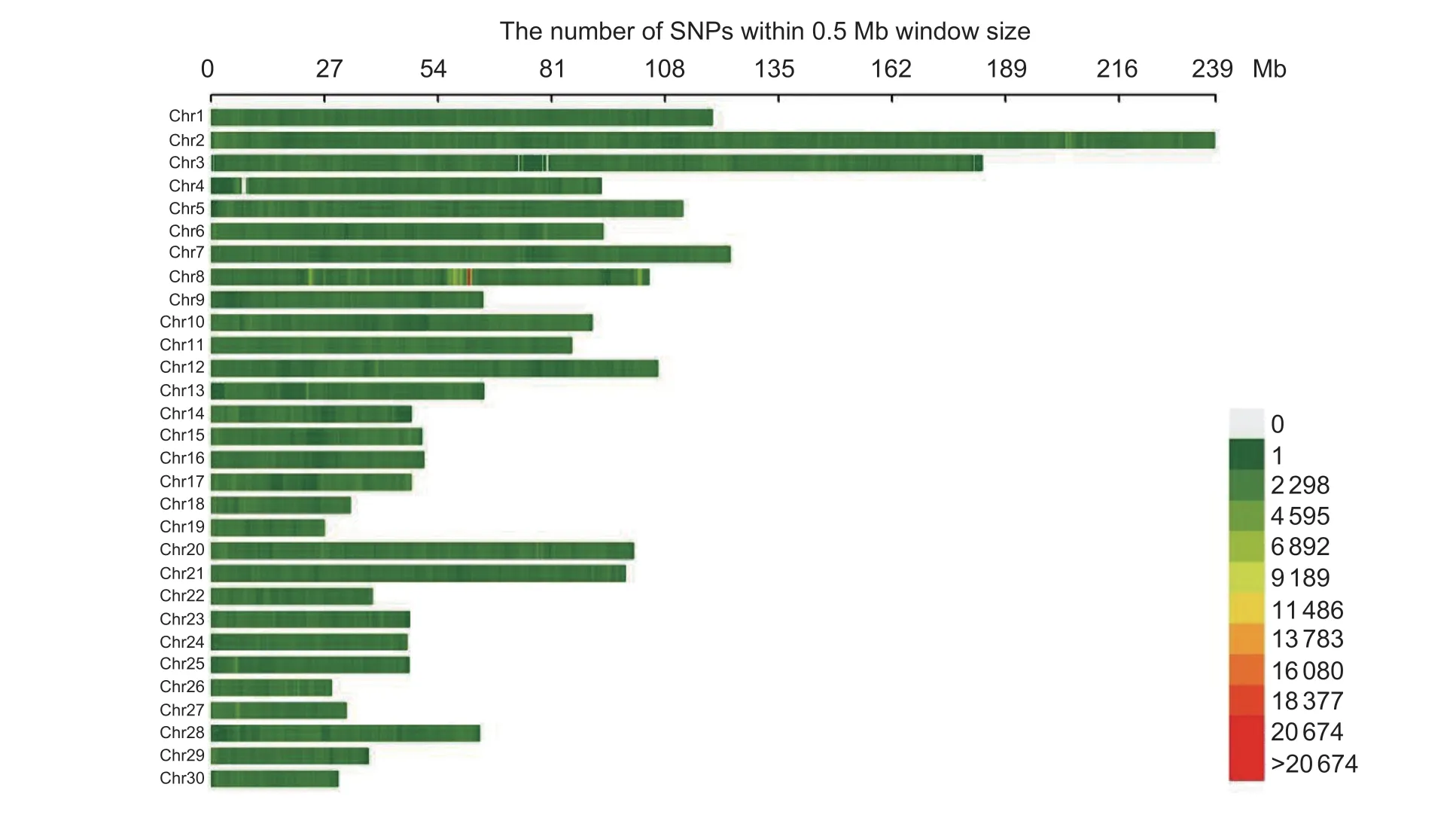
Fig.2 SNPs distributions on each chromosome after quality control.

Table 1 Numbers of animals with different numbers of thoracic and lumbar vertebrae
3.4.Population stratification analysis
The population stratification was evaluated on the basis of genome wide SNP data of autosome. PCA indicated that there was indeed stratification in the study population,the first PC divided the population into 2 groups and the second PC further divided one of them into 2 subgroups,with only a few outliers (Fig.3). The first PC explained 20.42% of the variation and the second PC explained 14.08% of the variation. Therefore,the first 2 PCs were used as covariables in the GWAS models to account for the population stratification effect.
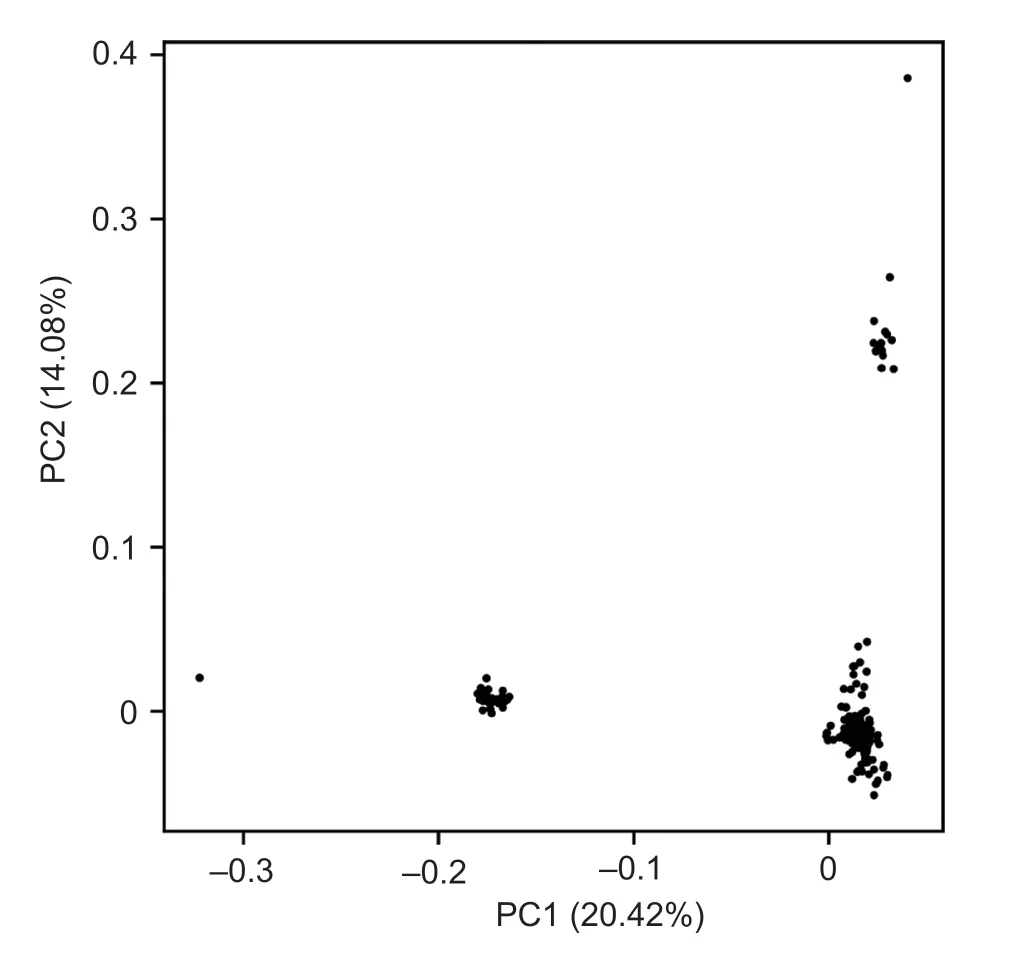
Fig.3 Principle component analysis of population using genome wide SNPs data.
3.5.Significant SNPs and candidate genes for TVn
The Manhattan plots and Q–Q plots of GWAS results based on linear and logistic models are shown in Fig.4.The Q–Q plots indicated that the GWAS results should not suffer from population stratification by including principle components in the models. Moreover,the inflation factors (λ) for TVn and LVn were 1.030 and 1.019,respectively,using linear mixed model;and 1.020 and 1.009,respectively,using logistic mixed model,suggesting that the effect of population stratification was effectively removed after including the first two PCs in the models. Two significance levels were considered: 5%genome-wide significance withP≤0.05/353 672=1.41e–07 and suggestive significance withP≤1/353 672=2.83e–06.Using the linear mixed model,a total of 167 SNPs was identified to be significant at the suggestive significance level (Fig.4-A;Appendix A),among which 38 SNPs on Chr5,21,and 23 were also significant at the genomewide significance level. Using the logistic mixed model,77 SNPs were found to be significant at the suggestive significance level (Fig.4-B;Appendix B),among which 26 were overlapped with those from the linear mixed model,but no SNPs reached genome-wide significance level.We focused on the SNPs of genome-wide significance and the SNPs of suggestive significance shared by the 2 models (64 in total,Appendix C). These SNPs are primarily located on Chr5 (14 SNPs),Chr12 (20 SNPs),and Chr23 (28 SNPs),only 2 are located on Chr7 and Chr21,respectively.
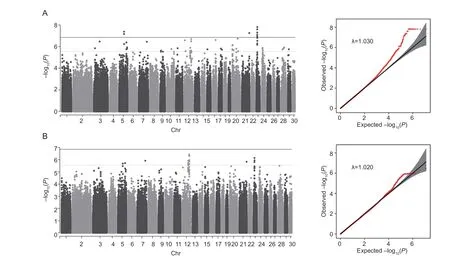
Fig.4 Manhattan plot and quantile-quantile plot for the number of thoracic vertebrae (TVn) using 2 models. A,the results of linear mixed model. B,the results of logistic mixed model. The dotted line indicates suggestively significant threshold (P=2.83e–06) and the solid line indicates 5% genome-wide significance threshold (P=1.41e–07).
Of the 14 SNPs on Chr5,10 were genome-wide significant and 4 were suggestive significant. The 10 SNPs are located within a <5-kb region in introns of theNLGN1(neuroligin 1) gene. The 4 suggestive SNPs are in upstream (80.2–82.1 kb) ofLOC123286078. On Chr12,all of the 20 SNPs were suggestive significant. The 17 of them are located in introns of theTOX(thymocyte selection-associated high mobility group box factor) gene,and the other 3 are located betweenRUNX1T1(RUNX1 partner transcriptional co-repressor 1) andSLC26A7(solute carrier family 26 member 7),about 60.6–92.6 kb away from the downstream ofSLC26A7. On Chr23,all of the 28 SNPs were genome-wide significant. A total of 22 of them are located in the upstream (0.4–60.4 kb) and 6 in introns ofLOC123280142. The one SNP on Chr7 is located in an intron ofDCC(DCC netrin 1 receptor) and the one on Chr21 is located in an intron ofWNT7A(Wnt family member 7A).
Since most of the significant SNPs on Chr5,Chr12,and Chr23 are closely linked with each other,we performed haplotype block analysis for these SNPs. The 14 SNPs on Chr5 were divided into 2 blocks,the 10 GWS SNPs withinNLGN1in one block and the 4 SNPs near toLOC123286078in another.within the 2 blocks were all equal to 1,indicating perfect LD. The 20 SNPs on Chr12 were also divided into 2 blocks,the 3 SNPs near toSLC26A7in one block and the 17 SNPs withinTOXin another. Again,between SNPs within blocks were nearly 1 (=0.97 except one with 0.91). The 28 SNPs on Chr23 are all in one block,withequal to 0.91–1.0(Appendix D).
3.6.Significant SNPs and candidate genes for LVn
The GWAS results for the number of lumbar vertebrae is illustrated in Fig.5. The same suggestive and genomewide significance levels as used for TVn were also applied for LVn. Based on the linear mixed model,we identified 107 SNPs of suggestive significance,of which 9 were reached genome-wide significance. Based on the logistic model,47 SNPs were identified to be suggestively significant,of which 4 on Chr5 were of genome-wide significance,and all of these SNPs appeared in the result of the linear mixed model. Details of these SNPs and genes related to them are given in Appendix E. Again,we focused on the genome-wide significant SNPs and suggestively significant SNPs commonly identified by the 2 models. They are located on Chr10 (27 SNPs),Chr15(15 SNPs),Chr20 (3 SNPs),Chr26 (1 SNP),and Chr29 (1 SNP).
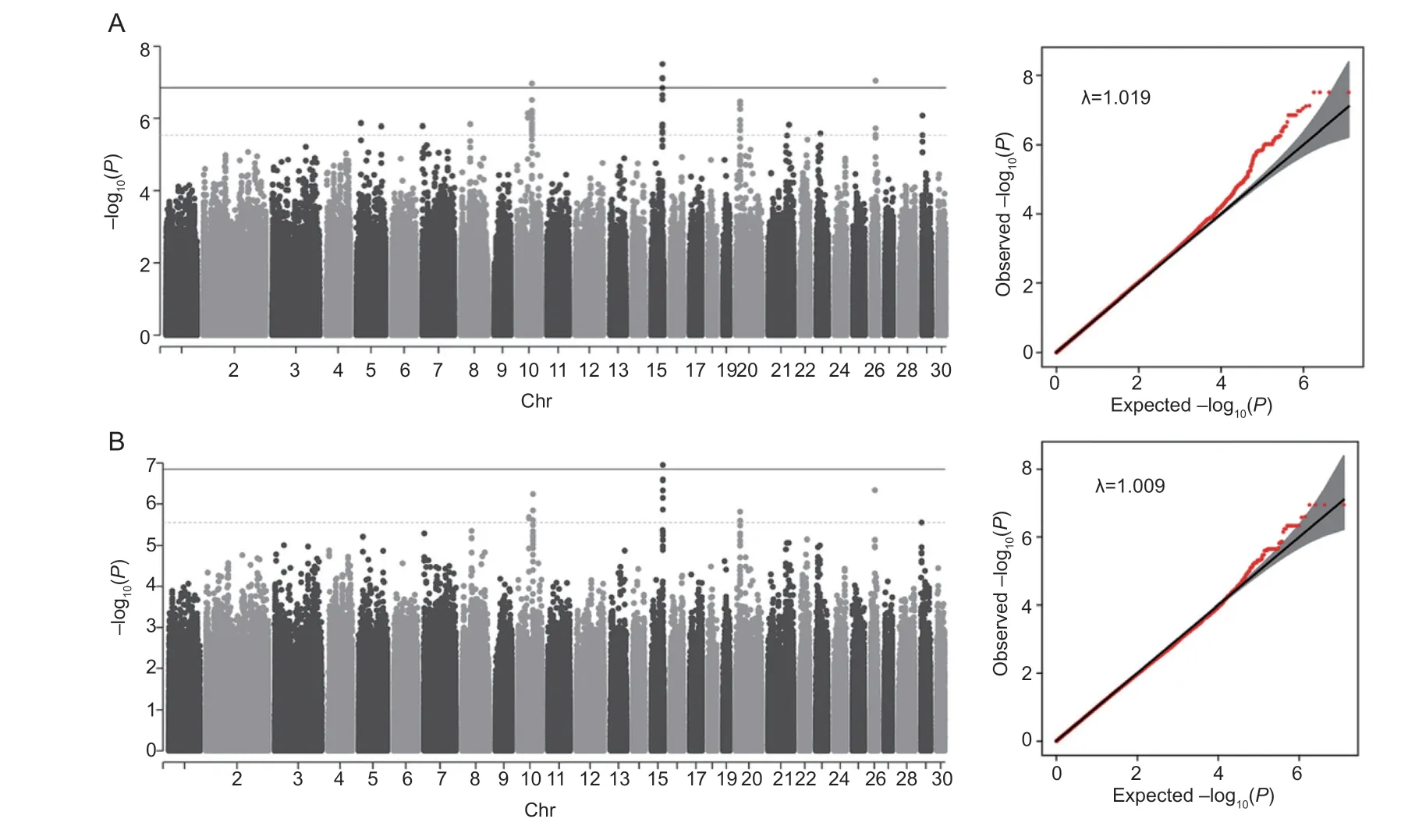
Fig.5 Manhattan plot and quantile-quantile plot for the number of lumbar vertebrae (LVn) using 2 models. A,the results of linear mixed model. B,the results of logistic mixed model. The dotted line indicated suggestively significant threshold (P=2.83e–06)and the solid line indicated 5% genome-wide significance threshold (P=1.41e–07).
Among the 27 SNPs on Chr10,18 are in introns ofGABBR2(gamma-aminobutyric acid type B receptor subunit 2),the other 9 are in the downstream (109–138 kb)ofFBXO4(F-box protein 4). Among the 15 SNPs on Chr15,4 are in intron ofLOC123277146,2 and 7 in the downstream (43 bp and 1 kb) and upstream (0.9–4.7 kb),respectively,ofLOC123277359,and 2 in the downstream(73.8 kb) ofBMP7(bone mprphogenetic protein 7). The 3 SNPs on Chr20 are in the upstream (6–36 kb) ofB3GAT1(beta-1,3-glucuronyltransferase 1). The SNP on Chr26 is in an intron ofEML2(echinoderm microtubule associated protein like 2). The SNP on Chr29 is in an intron ofLRP5(low density lipoprotein receptor related protein 5).
Haplotype block analysis showed that the 27 SNPs on Chr10 were divided into 2 blocks,the 18 SNPs withinGABBR2in one block and the 9 SNPs in the downstream ofFBXO4in another.within the two blocks were all equal to 1,indicating perfect LD. The 15 SNPs on Chr15 were also divided into 2 blocks,the 2 SNPs in the downstream ofBMP7in one block and the 13 SNPs within or close toLOC123277359in another. Again,between SNPs within blocks were equal to or nearly 1 (0.97–1).The 2 SNPs on Chr20 were in one block,withequal to 1 (Appendix F).
4.Discussion
Genome-wide association study can offer us new insights into the genetic basis of economic traits. Low-coverage whole genome sequencing followed by imputation has been proved a feasible strategy for obtaining whole genome sequence data for GWAS (Nicodet al.2016;Yang Ret al.2021). In this study,linear mixed model and logistic mixed model were implemented to analyze association between whole genome SNPs (obtained by imputation of low-coverage sequence data) and TVn and LVn in Dezhou donkey. Population stratification and relationship among individuals were considered in both models. For TVn,we identified 38 genomewide significant SNPs and 64 suggestive significant SNPs (revealed by both models),which involve 7 genes(NLGN1,DCC,SLC26A7,TOX,WNT7A,LOC123286078,andLOC123280142). For LVn,we identified 9 genomewide significant SNPs and 38 suggestive significant SNPs,which involve 8 genes (GABBR2,FBXO4,LOC123277146,LOC123277359,BMP7,B3GAT1,EML2,andLRP5).
Associations between genomic variants and number of vertebrae in pigs were investigated extensively by using chip-based GWAS and a number of genes were reported to be strongly associated with TVn,LVn,or TVn+LVn. In particular,theVRTN(Vertnin) gene was identified to be associated with TVn or TVn+LVn by several studies in different populations (breeds) (Mikawaet al.2011;Fanet al.2013;Rohreret al.2015;Zhanget al.2016;Duanet al.2018;Niuet al.2021). Moreover,this gene was confirmed to be associated with TVn in sheep (Liet al.2019). In addition toVRTN,the reported genes includeNR6A1(an orphan nuclear receptor,germ cell nuclear factor) for TVn+LVn (Mikawaet al.2007),HOXAandHOXBfor TVn and LVn,respectively (Rohreret al.2015),andABCD4for TVn+LVn (Niuet al.2021). Unexpectedly,these genes were not confirmed in our study in donkey.We speculate that the genetic mechanism for number of vertebrae might be somehow different between pigs and donkey,or in general,between artiodactyl and in perissodactyla animals.
Among the genes we revealed for TVn and LVn,3 genes,WNT7A,BMP7andLRP5attracted our attention because of their known functions.WNT7Ais a member of the WNT gene family. During embryonic development,the WNT signaling activates the canonical Wnt/β-catenin pathway and promotes the expression of posterior axis genes (Hikasa and Sokol 2013).WNT7Aplays an important role in the transcriptional response to β-catenin signaling for mammalian limb development through its synergy with theTBX3gene (Zimmerliet al.2020) and loss ofWNT7Awould produce defects in limb development (Wanget al.2022). A GWAS of skeletal phenotypes showed that many members in the WNT pathway were associated with bone mineral density(Hsu and Kiel 2012). Wnt/β-catenin signaling pathway also plays a crucial role in the regulation of osteogenic differentiation of bone mesenchymal stem cells (BMSCs)(Xuet al.2019). Yang Let al.(2021) found thatWNT7Aplays a key role in BMSCs differentiation into osteoblasts by regulating theRUNX2gene,and the RUNX2 protein is essential for skeletal morphogenesis. In the early somite stage of mouse embryos,WNT7Ais a dorsalizong signal to control dorsal-to-ventral transformations of cell fate (Parr and Mcmahon 1995). In pigs,althoughWNT7Awas not identified as a candidate gene for number of vertebrae,but Rohreret al.(2015) found that its family membersWNT7BandWNT5Bwere significantly associated with TVn.
BMP7belongs to the transforming growth factor-β(TGF-β) superfamily,which mediates osteoblastic differentiation and induces osteoblast proliferation(Laveryet al.2009).BMP7may exert functions in bone development (Chenet al.2021). In mice,Jenaet al.(1997) found that theBMP7homozygous null mice could lead to postnatal lethality and the vertebral column presented various defects,including lack of one lumbar vertebra. In addition,there is cross talk between Wnt and TGF pathways during axis specification (Hikasa and Sokol 2013). During murine embryos development,BMP7was detected in tissues that are known to be the source of inductive signals,may mediate aspects of interactions between these tissues (Lyonset al.1995). In addition,a homologue gene ofBMP7,BMP6was found to be significantly associated with thoracolumbar variation in pigs (Rohreret al.2015).
LRP5is essential in bone development (Freyet al.2015;Heet al.2020). The specificity of LRP5 bind several different molecules,principally the Wnt protein,modulation β-catenin signaling pathway in mechanical loading-induced bone formation (Kang and Robling 2015).Katoet al.(2002) emphasized thatLRP5is required for normal proliferation and function of osteoblasts. Yadavet al.(2010) found thatLRP5may act in gut cells to control bone formationviaa Wnt-independent pathway and regulate bone mass. The mutations ofLRP5cause the bone defects,suggesting the critical role of this gene in skeletal integrity (Gonget al.2001). The mice with deficiency inLRP5have been reported to have low bone mass,low body weight,and abnormal eye vascularization(Levasseuret al.2001). Conditional knockout (CKO) mice ofLRP5also presented reduced body size and shortened limb due to impaired bone formation (Linet al.2021). In human,multiple SNP mutations inLRP5were found to be associated with bone mass variation disease,including high bone mass,osteopetrosis,and osteoporosis (Littleet al.2002;Levasseuret al.2005;Xuet al.2014).LRP8,another member of the LRP family,was found to be significantly associated with TVn variation in swine (Rohreret al.2015).
5.Conclusion
A GWAS using imputed whole sequence data was carried out to detect the associations between genomic variants and TVn and LVn in Dezhou donkey. For TVn,we identified 38 genome-wide significant and 64 suggestive SNPs,which relate to 7 genes (NLGN1,DCC,SLC26A7,TOX,WNT7A,LOC123286078,andLOC123280142).For LVn,we identified 9 genome-wide significant and 38 suggestive SNPs,which relate to 8 genes (GABBR2,FBXO4,LOC123277146,LOC123277359,BMP7,B3GAT1,EML2,andLRP5). The genes (WNT7A,BMP7,andLRP5) involve in the Wnt and TGF-β signaling pathways and may play important role in embryonic development or bone formation and could be good candidate genes for TVn and LVn.
Acknowledgements
This study was supported by the Natural Science Foundation of Shandong Province,China(ZR2020MC168). We thank Dong-E E-Jiao Co.,Ltd.(Liaocheng) for supporting collecting phenotype data and blood samples.
Declaration of competing interest
The authors declare that they have no conflict of interest.
Ethical approval
All procedures involving animals were approved by the Institutional Animal Care and Use Committee at Shandong Agricultural University and were carried out in accordance with the guiding principles for the care and use of laboratory animals (SDAUA-2018-018).
Appendicesassociated with this paper are available on https://doi.org/10.1016/j.jia.2023.04.038
杂志排行

Journal of Integrative Agriculture的其它文章
- The association between the risk of diabetes and white rice consumption in China: Existing knowledge and new research directions from the crop perspective
- Linking atmospheric emission and deposition to accumulation of soil cadmium in the Middle-Lower Yangtze Plain,China
- Are vulnerable farmers more easily influenced? Heterogeneous effects of lnternet use on the adoption of integrated pest management
- lnfluences of large-scale farming on carbon emissions from cropping:Evidence from China
- Spatio-temporal variations in trends of vegetation and drought changes in relation to climate variability from 1982 to 2019 based on remote sensing data from East Asia
- Optimizing water management practice to increase potato yield and water use efficiency in North China