PPy对Al掺杂MnO2电极材料电化学性能的影响
2023-10-16李佳萌曾啸雄谈国强
夏 傲, 李佳萌, 曾啸雄, 陈 军, 谈国强
(陕西科技大学 材料科学与工程学院 陕西省无机材料绿色制备与功能化重点实验室, 陕西 西安 710021)
0 引言
超级电容器是一种电化学能源,是基于电化学能量转换原理的非常规能源设备,其具有比电容高、充放电速度快、循环寿命长、稳定性好和环境友好等优点,有广阔的应用前景,备受研究人员关注[1-4].
超级电容器的性能受到很多因素的影响,例如电极材料的电化学性能、电解质的选择以及电极的电位窗口,电极材料的选择在其中至关重要[5-8].因此,大量的研究工作致力于开发具有适当结构设计的超级电容器电极先进材料,以促进有效的电子传输和离子扩散[9].二氧化锰(MnO2)具有低成本、高比电容和环境兼容性,被广泛用于电极材料,但作为半导体材料,MnO2的低电导率和低离子传输效率等缺点限制了它的实用性[10-13].
为了解决上述问题,研究人员提出了几种改性方法:(1)提高电极的导电性[14];(2)使用纳米结构,最大限度地减少扩散途径,同时获得更高的比表面积[15];(3)开发多孔材料以加速离子扩散[16].其中,向MnO2中掺杂金属离子Al是一种有效的改性方法.Hu等[17]采用水热法制备了Al掺杂的α-MnO2电极,比电容达到213 F/g,在15 000次循环后还能保持91%的循环稳定性.Huang等[18]通过化学沉淀方法合成了Al掺杂的MnO2,在1 A/g的电流密度下比电容为264.6 F/g,高温比电容可达325.4 F/g,在室温和50 ℃下都表现出良好的循环稳定性.此外,夏傲等[19]采用水热法制备Al3+离子掺杂δ-MnO2,在1 A/g的电流密度下Al3+掺杂量为45%的电极(A0.45M)比电容为207.61 F/g,循环10 000次后比电容保持率为81.33%.单独的离子掺杂虽然可以提高电极材料的电导率,但结构不够稳定,MnO2易发生体积效应的问题还未得到解决.
聚吡咯(PPy)由于其良好的本征电导性和经济性,在超级电容器中显示出很好的应用前景,由于其快速的氧化还原反应、生物相容性以及较高的机械强度,在提高电化学性能方面备受关注[20,21].所以,通过复合材料设计将MnO2与PPy进行结合不仅可以提高超级电容器的电导率,还可以减小电极材料的体积效应.Wang等[22]通过模板法制备了MnO2@PPy复合材料,在1 A/g的电流密度下的比电容为295 F/g.组成的非对称电容器在1 A/g的电流密度和0~2.2 V的电压窗口下具有63 F/g的比电容,42 Wh/kg的能量密度和1 100 W/kg的功率密度.Zhou等[23]通过原位聚合法制备了核壳结构的PPy@MnO2复合材料,低电流密度下的比电容为614.7 F/g.与碳组成的非对称电容器具有0~1.6 V的电压窗口,34 Wh/kg的能量密度和317.6 W/kg的功率密度.然而,对于δ-MnO2先进行离子掺杂再与PPy复合的报道很少.
因此,本课题通过水热法和低温原位聚合法,制备Al3+掺杂MnO2与PPy的复合材料,研究复合量的变化对MnO2电极电化学性能的影响规律,得到了最佳的复合量,并深入分析PPy复合对δ-MnO2电化学性能提高的机理.
1 实验部分
1.1 主要原料
高锰酸钾(KMnO4),国药集团化学试剂;盐酸(HCl),国药集团化学试剂;一水合硫酸锰(MnSO4·H2O),国药集团化学试剂;六水合氯化铝(AlCl3·6H2O),阿拉丁化学试剂;乙醇(C2H5OH),国药集团化学试剂.吡咯(C4H5N),国药集团化学试剂.泡沫镍(Ni),云纵城科技有限公司.
1.2 Al掺杂MnO2的制备
用水热法制备Al掺杂的MnO2.称取0.19 g KMnO4、0.034 g MnSO4·H2O和0.022 g AlCl3·6H2O,溶于40 mL去离子水中.搅拌均匀后将溶液移入聚四氟乙烯内衬的高压反应釜,在150 ℃下保温16 h.反应结束后收集并洗涤产物,在70 ℃下干燥12 h,得到产物,命名为A0.45M.在本课题组之前的研究中,已对Al的掺杂量进行ICP-AES测试,得到A0.45M中实际掺杂量(摩尔分数)为0.56%[19].
1.3 Al掺杂MnO2复合PPy的制备
称取一定量的A0.45M(0.270、0.231、0.193、0.154 g),量取20 mL浓度为0.1 mol/L的盐酸,混合超声30 min后加入0.04 mL吡咯单体(Py),搅拌至溶液颜色变黑,移入冰水浴中在0 ℃下反应4 h.将反映后的物质洗涤干净,再置入70 ℃干燥箱干燥12 h,得到A0.45M/PPy复合材料.根据Py的用量将样品分别记为A0.45M7P1、A0.45M6P1、A0.45M5P1、A0.45M4P1(A0.45M与Py的质量比分别为7∶1、6∶1、5∶1、4∶1).
1.4 样品的形貌和结构表征
采用X射线衍射仪(XRD,D/max2200PC,日本理学株式会社)对样品进行物相分析,Cu靶Kα为辐射源,波长λ=0.154 06 nm,管电压40 KV,管电流100 mA,扫描角度10°~80°,扫速20°·min-1.利用场发射扫描电子显微镜(SU8100,日本HITACHI公司)与透射电子显微镜(TEM,FEI Tecnai G2 F20 S-TWIN,美国)对样品进行形貌和微观结构表征.通过傅里叶红外光谱测试(FT-IR,VERTEX-70,德国)确定样品的分子结构以及化学组成,测试波长为400~4 000 cm-1.通过热重分析测试(TG,TGA Q500,美国)获取材料组分的含量以及热稳定性,升温速率为10 ℃/min,升温范围为40 ℃~650 ℃.
1.5 电极的制备及电化学性能分析
称取35 mg的活性物质,10 mg的导电碳粉,5 mg的PVDF(聚偏氟乙烯),依次放入玛瑙研钵中充分混合研磨半小时后,加入适量分散剂NMP(N-甲基吡咯烷酮)充分研磨至具有一定粘度的浆料.将其均匀涂覆在裁剪成1×2 cm大小的泡沫镍上,放入真空干燥箱中,70 ℃下干燥24 h.涂膜前与干燥后称量记录电极片,计算活性物质的量(活性物质负载量为0.85~1.00 mg·cm-2).
恒流充放电性能测试(GCD,0~1 V,1~10 A/g)、循环伏安测试(CV,0~1 V,5 mV/s)以及电化学阻抗测试(EIS,0.001~100 000 Hz)由电化学工作站(CHI 660E)完成.
2 结果与讨论
2.1 XRD分析
图1是A0.45M以及在其表面包裹了有机导电聚合物吡咯(PPy)的A0.45M/PPy复合材料样品的XRD谱图.从图中可以看出,包裹PPy后,样品的XRD谱图相较于A0.45M没有发生明显变化,只是衍射峰强度有所下降.并且随着PPy复合量的增加,衍射峰的强度逐渐降低.
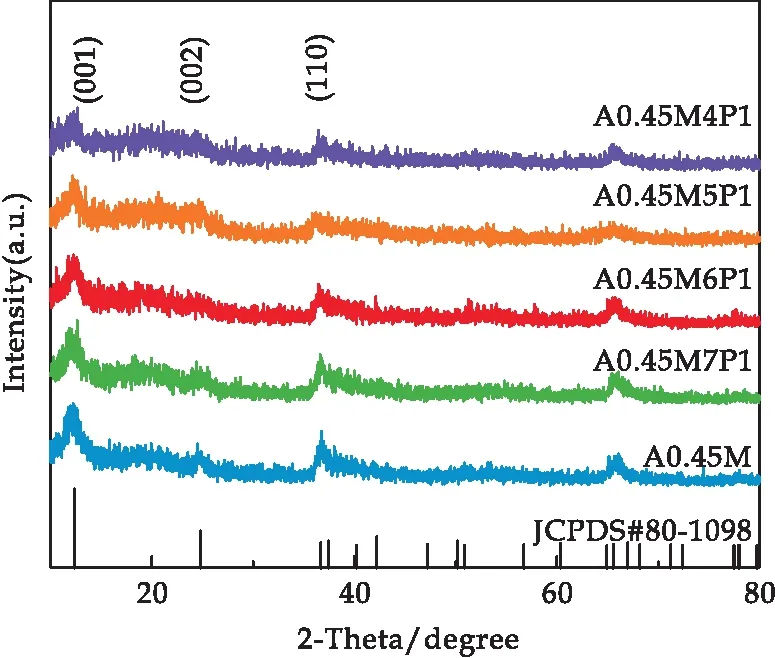
图1 所有样品的X射线衍射图
2.2 SEM分析
图2是A0.45M以及在其表面包裹了高分子导电聚合物吡咯(PPy)的A0.45M/PPy复合材料样品的SEM照片.图2(b)、(g)为A0.45M7P1的SEM照片,发现和A0.45M对应的图2(a)、(f)SEM照片没明显变化,只是纳米片的的厚度由12 nm增加到了16 nm,这可能是加入PPy较小,导致PPy对A0.45M包覆效果不明显.

图2 样品的SEM图
图2(c)、(h)是A0.45M6P1的SEM照片,随着PPy复合量的增大可以看出纳米片明显变厚,纳米片的厚度达到22 nm并且花球孔洞有明显的填充;图2(d)、(i)为A0.45M5P1的SEM照片,随着PPy复合量的进一步加大,可以看到纳米片的厚度并没有继续增大依旧保持22 nm,但是A0.45M5P1花球组成的团簇发生了明显的团聚,花球内部的空隙大小明显变小;图2(e)、(j)为 A0.45M4P1的SEM图片,A0.45M4P1呈现的形貌与前几种样品都有所不同,产生了大量实心球和小部分花球,花球内部空隙也逐渐被填满,导致了严重的团聚现象.团聚现象致使复合材料比表面积减小,活性位点随之减少,导致性能下降.
2.3 TEM分析
TEM照片进一步揭示了样品A0.45M6P1的微观形貌.由图3(a)可以看出,A0.45M6P1样品还是成花球状,花球直径依旧为310 nm,但是花球之间的空隙相较于A0.45M变得更窄了.这主要是因为PPy的加入,与A0.45M完成了复合,PPy包裹住了表面的纳米片,从而导致纳米花球之间的空隙被填充.图3(b)是高压下TEM照片.通过EDS能谱表明,A0.45M6P1样品中含有Mn、O、K、Al、C和N元素(图3(c)~(h)).K离子在水热过程中对δ-MnO2层状结构的形成有着稳定结构的作用,对电化学反应无影响[24].C和N元素的均匀分布预示了PPy成功地与A0.45M完成了复合.
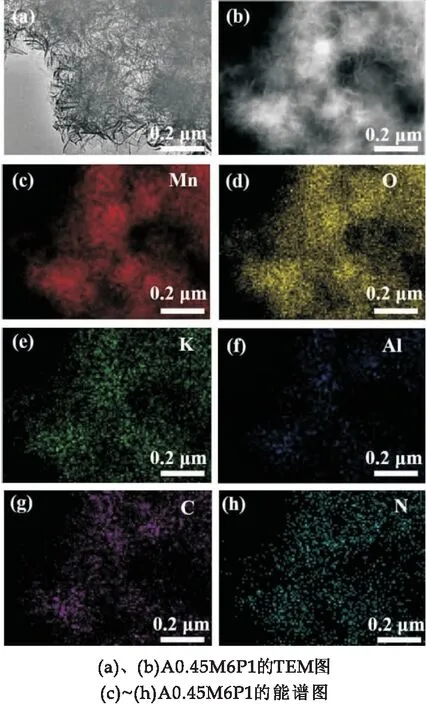
图3 样品的TEM图
2.4 傅里叶红外光谱分析(FT-IR)
采用FT-IR对A0.45M和A0.45M6P1复合材料进行化学键结构分析测试,其结果如图4所示.其中,464 cm-1、510 cm-1、799 cm-1处出现的特征峰来源于MnO2中[MnO6]的Mn-O键伸缩振动[25,26].在1 010 cm-1、1 060 cm-1、1 093 cm-1和1 170 cm-1处出现的特征峰分别对应于=C-H键面外伸缩振动、N-H键面内伸缩振动、C-N键伸缩振动和=C-H键面内伸缩振动[23,25].1 400 cm-1、1 630 cm-1、1 740 cm-1处对应的特征峰分别对应于C-C键的伸缩振动、C=C键面内伸缩振动以及羰基/羧基相关的C=O键伸缩振动,而1 740 cm-1处出现的特征峰表明PPy与被包裹物质之间存在明显的界面相互作用[23,26].经过对比可得PPy成功与A0.45M完成了复合.
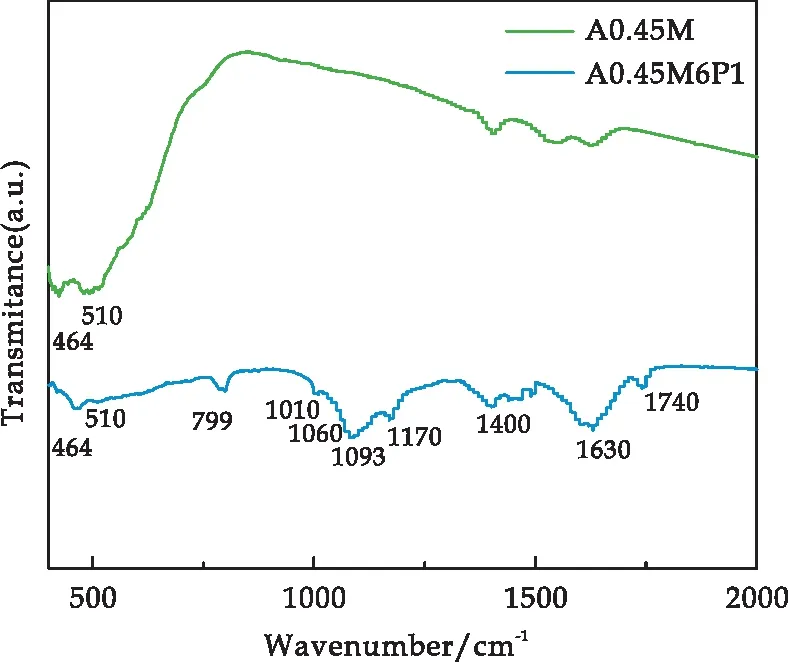
图4 A0.45M和A0.45M6P1样品的红外光谱
2.5 热重分析(TGA)
为确定A0.45M6P1复合材料中各组元的含量,对样品进行TGA测试,其结果如图5所示.在空气气氛下进行测试,升温速率为10 ℃/min,升温范围为40 ℃~650 ℃.从图中可以了解到,TGA曲线大致可以分为三个阶段.第一个阶段(40 ℃~205 ℃):A0.45M6P1样品在205 ℃之前一共失重8.7%,该过程主要是为了除去样品表面的吸附水和内部的结晶水;第二个阶段(205 ℃~550 ℃):A0.45M6P1样品又发生了9%的失重,这个阶段主要是复合材料中的PPy的分子链受高温而产生了热分解;第三个阶段(550 ℃~600 ℃):该过程发生了很明显的失重现象,失重大小为1.69%,这主要对应MnO2的晶格失氧,Mn4+高温还原为Mn3+从而产生氧气先生成Mn2O3最终生成Mn3O4.TGA分析表明,A0.45M6P1复合材料中PPy的含量为9 wt%.
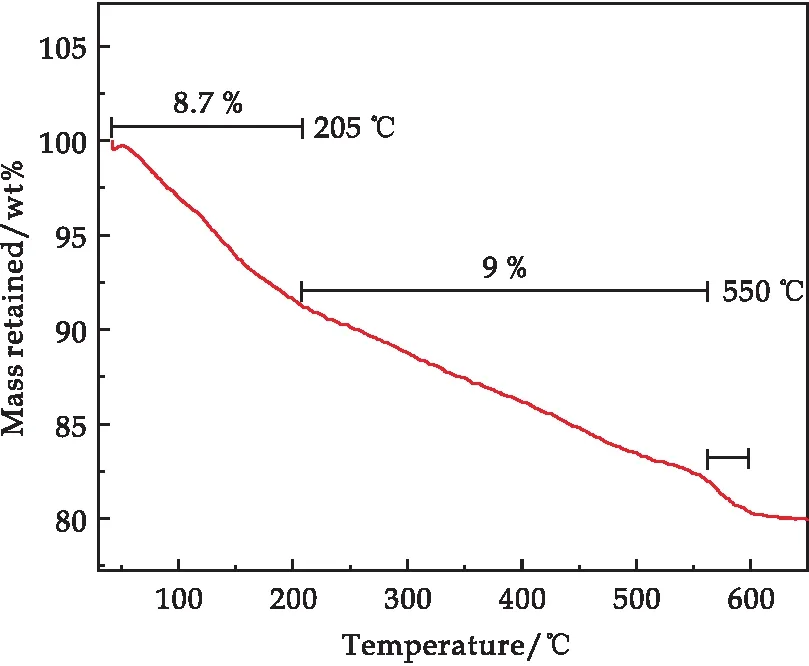
图5 A0.45M6P1复合样品的TGA谱图
2.6 电化学性能分析
图6(a)展示了不同样品在5 mV/s扫描速率下的CV曲线.由图可见,CV曲线呈矩形,表明电极具有理想的双电层电容特性和快速反应动力学特性.由A0.45M、A0.45M6P1在不同扫描速度下的CV曲线可以看出,即使在50 mV/s高扫描速率下,A0.45M、A0.45M6P1的CV曲线仍保持准矩形(图6(b)、(c)),说明样品具有良好的可逆性和超快的充放电能力.A0.45M6P1的CV曲线比A0.45M面积更大,这意味着PPy的复合提高了MnO2的反应动力学.
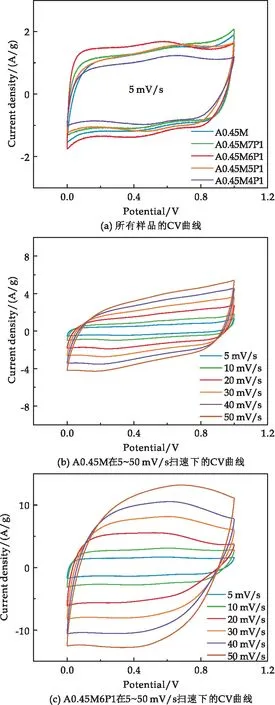
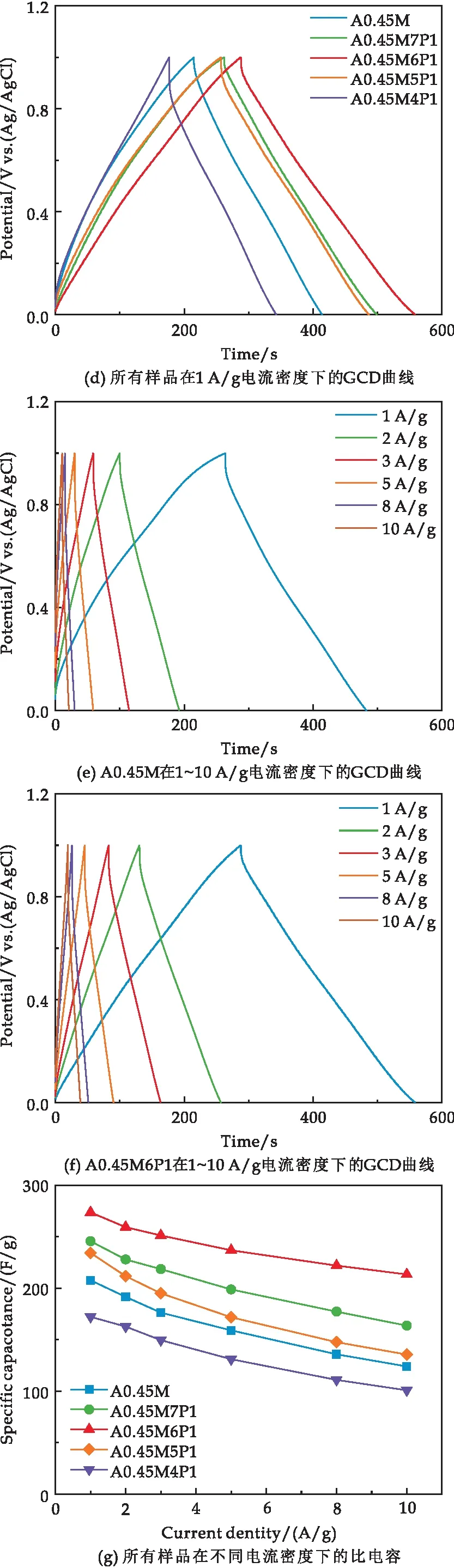
图6(d)为5种样品在1 A/g电流密度下的恒电流充放电曲线,所有曲线均呈等腰三角形.相比A0.45M,复合样品的的放电时间延长、内部电压(IR)降减小,其中A0.45M6P1的放电时间最长、IR降最小.IR降减小意味着样品反应极化减小,反应可逆性增强.在1 A/g电流密度下,A0.45M、A0.45M7P1、A0.45M6P1、A0.45M5P1和A0.45M4P1的比电容分别为207.61 F/g、245.62 F/g、277.56 F/g、234.22 F/g和172.15 F/g.
图6(e)、(f)分别为A0.45M和A0.45M6P1在1 A/g、2 A/g、3 A/g、5 A/g、8 A/g和10 A/g电流密度下的恒电流充放电曲线.随着电流密度的增加,A0.45M和A0.45M6P1的充放电时间均缩短.然而,在相同电流密度下A0.45M6P1充放电时间约为A0.45M的1.3倍,说明A0.45M6P1的倍率性能显著优于A0.45M.
图6(g)对比了所有样品在不同电流密度下的比电容.由图可知,随着电流密度(1、2、3、5、8、10 A/g)的增加,所有样品的比电容呈下降趋势,具体数值见表1所示.可知,相同电流密度下A0.45M6P1始终具有最高的比电容.电流密度从1 A/g增加至10 A/g时,A0.45M、A0.45M7P1、A0.45M6P1、A0.45M5P1和A0.45M4P1的比电容保持率分别为59.70%、66.60%、77.89%、57.84%和58.51%,对比可知适量的PPy复合可以有效提高MnO2的倍率性能,但过量的PPy会导致倍率性能下降,这与SEM的结果一致.

表1 所有样品在不同电流密度下的比电容
图6(h)为5种样品在10 A/g电流密度下的循环性能测试结果.由图可知,当PPy的复合量较低时,有助于A0.45M放电比电容的提升.随着PPy的复合量不断增大不仅不会提高A0.45M的放电比电容还会抑制A0.45M放电,降低A0.45M的放电比电容.其中A0.45M6P1的放电比电容最大.经过10 000次循环后,0.45M、A0.45M7P1、A0.45M6P1、A0.45M5P1和A0.45M4P1的放电比电容分别为100.81 F/g、153.62 F/g、196.78 F/g、109.57 F/g和65.07 F/g,比电容保持率分别为81.33%、94.33%、93.20%、82.80%和66.40%.PPy复合量较低时,对A0.45M的循环稳定性有显著的提升,说明了A0.45M6P1具有良好的循环稳定性.
A0.45M6P1具有良好的电化学性能主要归功于PPy为充放电过程中的电子提供了快速传输的路径,在MnO2的纳米片上复合上一层PPy有利于缓解MnO2在充放电过程中离子嵌入/脱出带来的体积效应.
图7(a)为所有样品的EIS谱图.在低频段,A0.45M7P1、A0.45M6P1比A0.45M电极的斜率更陡峭而A0.45M5P1和A0.45M4P1比A0.45M的斜率倾斜度小,表明小复合量的PPy复合电极具有更理想的电化学双电层电容行为,在电极表面具有更高的离子扩散速率和更低的扩散电阻(Zw),而大复合量的PPy会抑制电极的电化学性能.高频段的EIS曲线都呈半圆弧型,数据拟合得到样品A0.45M、A0.45M7P1、A0.45M6P1、A0.45M5P1和A0.45M4P1的Rct分别为2.28 Ω、1.94 Ω、0.69 Ω、2.73 Ω和5.93 Ω,可见低复合量的PPy可以降低A0.45M的Rct值,提高A0.45M的导电性.但是高复合量的PPy会增大A0.45M的Rct值,降低A0.45M的导电性.
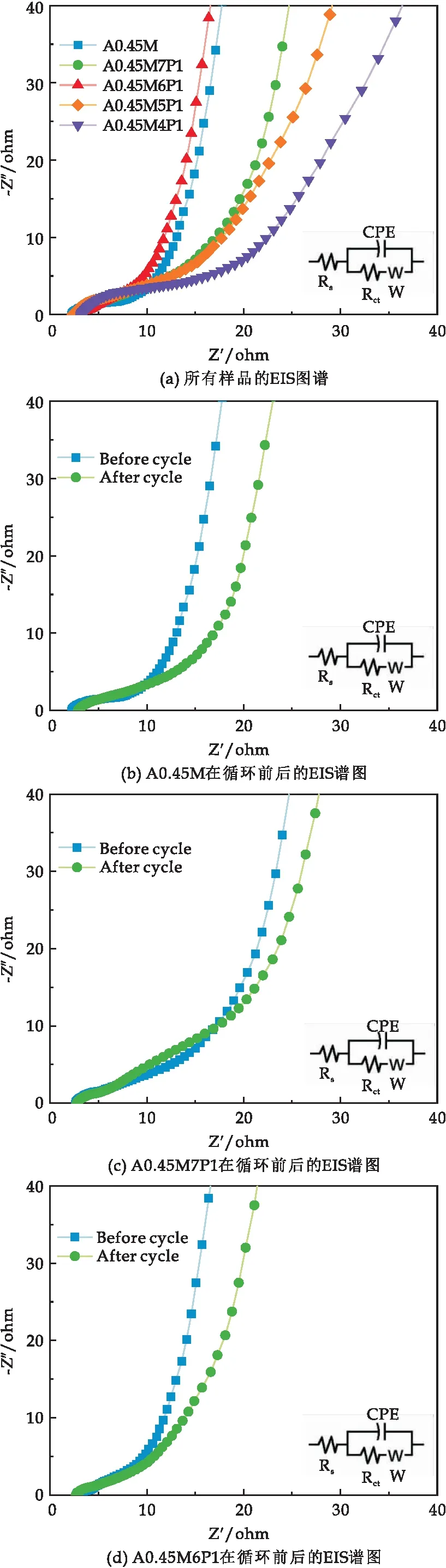

图7 样品的EIS谱图
图7(b)~(f)为5种样品循环5 000次前后的EIS图,分析得出所有样品循环前后的Rct值如表2所示.结果表明,低复合量PPy的A0.45M的Rct值循环前后都明显低于A0.45M,而高复合量PPy的A0.45M的 Rct值循环前后都高于A0.45M,这是因为在复合材料中,δ-MnO2作为支撑材料,为PPy的异质成核和均匀生长提供了丰富的活性位点,当PPy的复合量足够大时,会发生额外的自聚合反应,形成蠕虫状团聚体,反而会降低导电性[27].除此之外,A0.45M6P1循环前后低频区重合度最大,这说明了A0.45M6P1具有良好的结构稳定性.

表2 样品在循环前的EIS拟合数据以及在10 A/g的电流密度下循环5 000圈后的EIS拟合数据
在反应过程中PPy参与到电化学反应中,主要是由于PPy在Al掺杂的δ-MnO2(A0.45M)层状结构上可控生长,改善了A0.45M的导电性,与此同时A0.45M提供较大的电容性能,为我们优化复合材料的电容性能提供了机会.为进一步探究A0.45M6P1复合材料的电荷存储方式,根据A0.45M6P1在不同扫描速率(5~50 mV/s)的CV曲线(图6(c))进行分析.在CV图中,随着扫描速率的增加,阳极和阴极的峰值倾向于向正负方向扩大.阳极和阴极的峰值电流如图8(a)所示.峰值电流测量表明,电流响应与扫描速率线性相关[28].这不仅证明了电化学活性是由扩散控制的,而且在高扫描速率下也是可逆的[27].
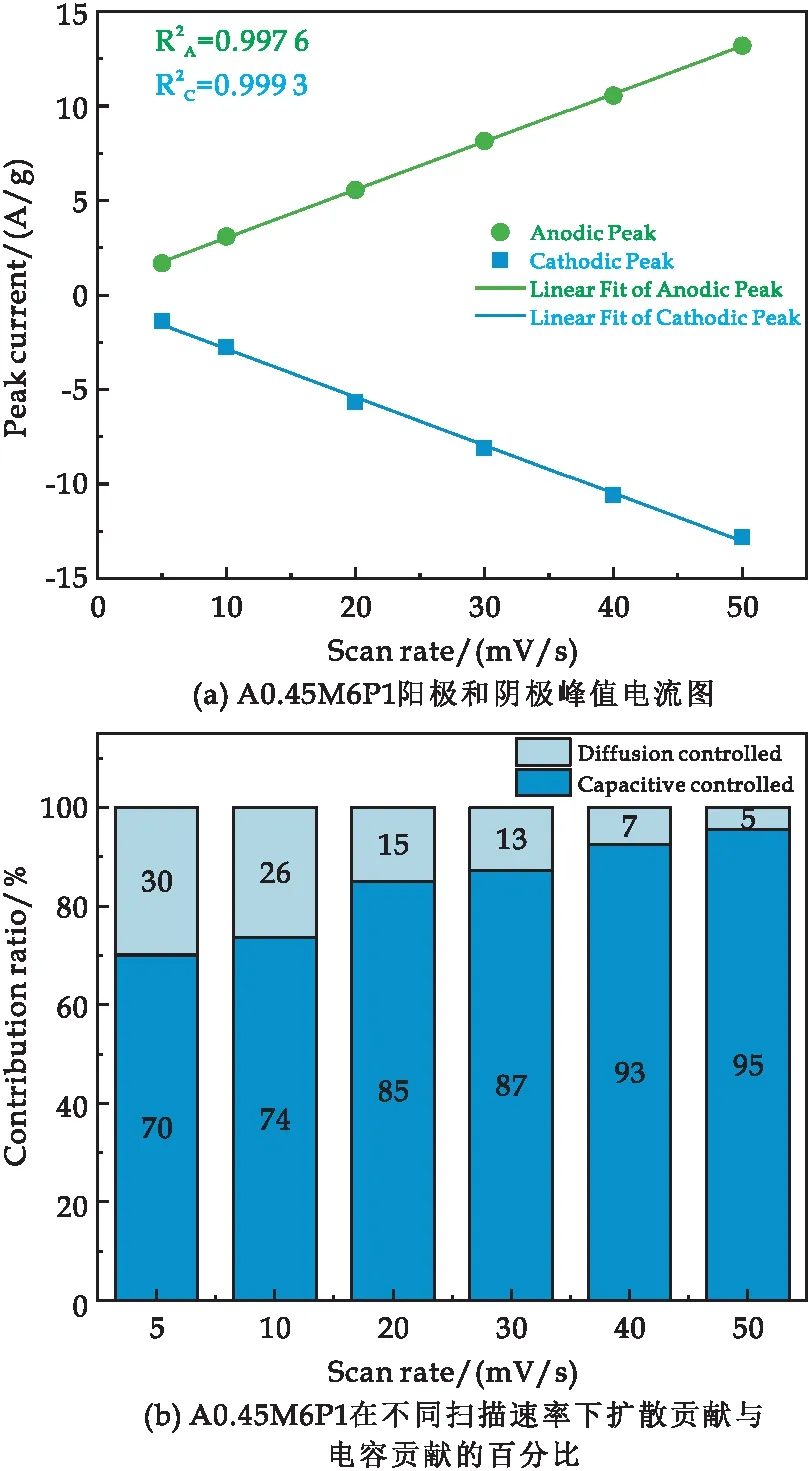
图8 A0.45M6P1样品的电化学性能图
赝电容贡献可按照公式(1)进行计算:
i(V)=k1v+k2v1/2
(1)
式(1)中:i为扫描速率为v时所得到的峰值电流大小,k1v代表了电容控制贡献的电流、k2v1/2代表了扩散控制贡献的电流.利用式(1)计算不同扫描速率下电容和扩散控制的贡献率如图8(b)所示.计算表明,当扫描速率较低时(5 mV/s),扩散控制贡献达30%,随着扫描速率的增加,电容控制贡献逐渐增大,说明电容控制占主导地位.
3 结论
本文以Al掺杂MnO2(A0.45M)电极材料为基础,通过低温原位聚合法制备了PPy复合A0.45M的电极材料,研究了不同复合量的PPy对A0.45M电极材料的组成、结构、性能的影响,并进一步了解PPy对A0.45M电化学性能作用的机理.
结果表明,相较于A0.45M样品,A0.45M6P1样品具有更好的电化学性能.PPy可以缓解MnO2在充放电过程中的体积效应,也可以提高MnO2的导电性.在1 A/g、2 A/g、3 A/g、5 A/g、8 A/g、10 A/g的电流密度下时A0.45M6P1比电容分别为273.56 F/g、259.22 F/g、251.19 F/g、236.78 F/g、221.93 F/g和213.34 F/g,电流密度从1 A/g增加至10 A/g时比电容保持率为77.89%,表明PPy可以进一步地提高A0.45M的电化学性能.
