N、S共掺杂煤基碳量子点的电化学氧化法制备及用于Fe3+检测
2023-10-14雷伟姜维佳王玉高和明豪申峻
雷伟,姜维佳,王玉高,和明豪,申峻
(1 太原理工大学化学工程与技术学院,山西 太原030024;2 山西中医药大学基础医学学院,山西 晋中 030619;3 山西大学环境科学研究所,山西 太原030006)
碳量子点具有光学稳定性高、发光强度高和毒性低等优势[1-3],因此在光催化[4]、生物传感[5]和药物递送[6]等新兴行业有着重要应用。自碳量子点被研发以来,其合成策略被不断革新,如尝试不同反应前体及改进反应方法来降低成本或优化性质[7]。相比于葡萄糖[8]、乙二胺四乙酸二钠(EDTA-2Na)[9]或有机化合物[10](如1,3-二羟基萘)等,煤尤其是褐煤作为反应前体有着更多优势。从经济角度来看,中国褐煤储量丰富且价格低廉;从结构角度来看,褐煤变质程度较低,挥发分高,其自身无序疏松的构型容易发生化学反应,并且低变质程度煤的结晶区域较小,是制备碳量子点的优良碳源[7,11]。2013 年,Ye 等[11]首次以煤作碳源,在浓硫酸和硝酸中超声处理2h,然后在120℃条件下反应24h,待产物冷却至室温,加入氢氧化钠中和,过滤并透析,得到碳量子点。但该法反应条件烦琐,荧光量子产率较低,且用到强酸强碱,会产生含盐废水。
为了提高荧光量子产率,常常需要对碳量子点进行掺杂[12],鉴于金属掺杂污染严重等问题,非金属元素掺杂得到广泛关注,其中N、S 掺杂最为常见。N元素掺杂可以通过引入新的表面态来提高荧光量子产率[13-15],而S 元素掺杂可以优化碳量子点的电子结构[16]。N、S共掺杂效果更优异,S元素的引入可以通过协同效应增强N掺杂碳量子点中N元素的诱导效应,并且引入更多的活性中心,提高荧光量子产率[17-19]。以上述机理为基础,Qie等[20]在二甲基甲酰胺中加入柠檬酸、硫脲,在高压釜中合成了N、S共掺杂的碳量子点,并成功用于检测Fe3+,但所用原料二甲基甲酰胺毒性较强,一定程度上限制了该方法的推广。
电化学氧化法可以制备碳量子点,且具有反应条件温和、绿色环保和方便控制等优势。传统电化学氧化法将煤制成碳电极,通电从电极上剥离出碳量子点,然而碳电极制备流程较为复杂,且无法重复利用[21]。本文将褐煤颗粒分散在氯化钠水溶液中,借助原位生成的次氯酸根离子氧化褐煤中的有机质,很好地解决了传统电化学氧化法中存在的问题。此外,将含N和S元素的掺杂助剂加入电化学氧化体系,制备了异质元素掺杂的碳量子点。
Fe3+广泛存在于生物体内及环境中,在生物化学系统中发挥着重要的生理作用,如促进细胞色素中的电子转移和组织中的氧传递等[22-24]。体内Fe3+缺乏会导致贫血,而血液中Fe3+浓度过高会导致肾脏损害和血色沉着,因此检测Fe3+浓度具有重要的意义[25]。传统的检测方法有紫外分光光度法和原子吸收法等,但这些方法因操作流程冗长或抗干扰能力差而在实际应用中受到限制。近些年荧光检测法得到广泛关注,与传统检测方式相比,荧光检测法具有检出限低、操作方便和抗干扰能力强等优势[26-29]。基于此,本工作利用所得的N、S共掺杂碳量子点(N,S-CQD)检测痕量Fe3+。
1 实验部分
1.1 试剂
氯化钠(分析纯,99.5%)、氢氧化钠(分析纯,96%)、磷酸氢二铵(分析纯,98%),天津市科密欧化学试剂开发中心;硫酸奎宁荧光标准物质(分析纯,99%)、硫脲(分析纯,99%),上海阿拉丁化学试剂有限公司;氨水(分析纯,28%),上海麦克林生化科技有限公司;乙二胺(分析纯,大于等于99.0%),国药集团化学试剂有限公司;云南昭通褐煤煤样(ZT),干燥处理,球磨机磨粉过200目筛后备用。
1.2 实验方法
1.2.1 碳量子点的制备
如图1 所示,将5g 氯化钠和0.1g 煤样溶于50mL 去离子水,然后通电进行反应,后续在混合物中分别加入氨水、乙二胺、硫脲或磷酸氢二铵等助剂,搅拌均匀后设定电流密度为0.5A/cm2,反应1.5h 后将得到的混合物在8000r/min 下高速离心,过滤残渣(FC1)得到滤液F1。调节滤液F1的pH至中性后再次过滤,将所得滤液F2旋蒸至50mL,装入透析袋,每日换水3次,透析3~5天,直至电导率小于200µS/cm2,收集透析袋内液体F3。取5mL用于表征,剩余液体减压蒸馏除水后,放入真空干燥箱干燥至恒重。

图1 掺杂碳量子点制备流程
1.2.2 碳量子点选择性检测痕量Fe3+
将5mL 碳量子点水溶液移入试管,加入适量Fe2(SO4)3后摇匀,使碳量子点溶液中Fe3+的最终浓度为150µmol/L。按以上步骤配制15种不同金属离子的同浓度碳量子点溶液,所有溶液均在室温下超声分散15min,在340nm 的激发波长下进行荧光测试。
1.3 碳量子点荧光量子产率的计算
利用参比法计算产物荧光量子产率(QY),以硫酸奎宁(溶于0.1mol/L H2SO4)作为具有已知荧光量子产率的标准参比物。通过测量标准参比样品和待测样品的荧光积分面积、吸光度等值,求得待测碳量子点的荧光量子产率。在实际测定过程中,高浓度会造成自吸收和内滤效应使荧光强度与浓度不成线性关系,因此在荧光测量中荧光物质的浓度不应太高,相应激发波长吸光度不超过0.1[30-31],如式(1)。
式中,Φst=0.54;Ix和Ist分别表示样品和参比溶液的荧光发射光谱的面积积分;Ax和Ast分别表示样品和参比溶液的吸光度值;nx和nst分别表示样品和参比溶液溶剂的折射率(n水=1.3330)。
1.4 碳量子点的表征分析
采用傅里叶变换红外光谱仪(FTIR,Nicolet iS50,Fisher Scientific 公司)对所得N,S-CQD 进行红外光谱表征,扫描范围为400~4000cm-1,使用Omnic 软件对FTIR 谱图校正处理;选用X 射线光电子能谱仪(XPS,ESCALAB 250XI,美国赛默飞世尔科技公司)对N,S-CQD进行XPS分析,X射线源为单色器为Al Kα,功率为150W,数据处理采用XPS PEAK41 软件;采用高分辨透射电子显微镜(TEM,JEM 2100F)观察N,S-CQD 表面形貌特征,加速电压为200kV,利用ImageJ软件测量粒径分布情况;采用紫外可见分光光度计(UV-1800,日本岛津有限公司)对N,S-CQD 水溶液进行紫外光谱分析,波长范围为190~1100nm,光谱带宽为2nm,最小取样间隔为0.1nm;选用荧光分光光度计(Hitachi F-2700,日立高新技术公司)对N,S-CQD水溶液进行荧光光谱分析,光电倍增管电压为400V,激发光和发射光的狭缝宽度均为10nm。
2 结果与讨论
2.1 褐煤的物性表征
表1为所选煤样的工业分析和元素分析,由表可知ZT 挥发分较高,氧含量高,而C/H 值较小,这证实了ZT 芳香度较小,芳环结构并不致密[32],其无序疏松的构型使得ZT容易发生化学反应。图2展示了ZT 的FTIR 谱图,3425cm-1处宽而强的吸收峰为—OH 的吸收峰,2920cm-1和2850cm-1处的特征吸收峰为C—H 的伸缩振动峰,1218cm-1处的吸收峰归属于C—O 的伸缩振动峰,1714cm-1处的吸收峰是C= = O 的伸缩振动峰,1613cm-1处的吸收峰是由芳环C= = C 骨架引起的。由FTIR 谱图可知ZT含有丰富的含氧官能团,其中—OH 和C= = O 等有助于提高褐煤氧化反应活性[7],这表明ZT是制备碳量子点的合适碳源。

表1 ZT的工业分析与元素分析(质量分数)

图2 ZT的FTIR谱图
2.2 助剂种类对碳量子点性能的影响
量子产率(QY)是碳量子点性质中基础且重要的参数,高QY表示碳量子点将吸收的光能更高效地转化为荧光,体现出更强的发光能力[33-34]。如图3 所示,以ZT 为碳源,利用电化学氧化法制备的碳量子点的QY 为0.90%,然后分别加入氨水、乙二胺、硫脲和磷酸氢二铵四种不同助剂,在相同反应条件下合成四种性能不同的碳量子点。为选取后续实验考察对象,对比四种助剂掺杂所制备碳量子点的QY,硫脲掺杂制备的碳量子点(N,S-CQD)的QY 为1.60%,优于其他三种助剂掺杂效果,所以后续以N,S-CQD作为实验对象。
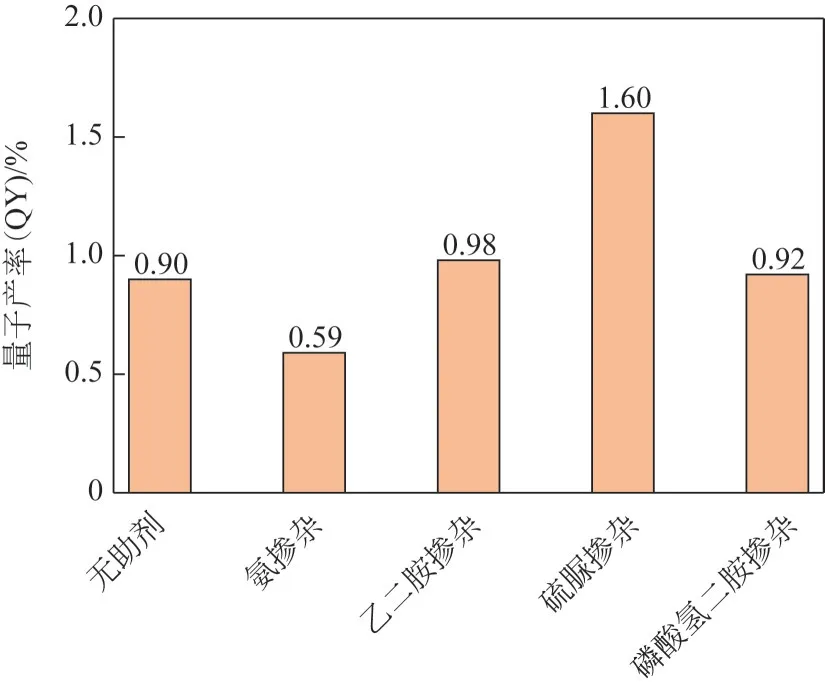
图3 无助剂及四种助剂掺杂后的碳量子点的QY对比
2.3 N,S-CQD的形貌、结构和组成分析
为了探究N,S-CQD 的尺寸及形貌特征,分析了N,S-CQD 的TEM 图以及尺寸分布。结果如图4所示,N,S-CQD 呈球形结构,整体分布较均匀。根据N,S-CQD 的尺寸分布图观察到N,S-CQD 的粒径为0.5~3nm,平均粒径为1.66nm,标准偏差为0.52nm,尺寸分布较窄。以上结果表明合成了纳米级别的碳量子点,有力地证明了电解氧化法合成碳量子点的可行性。
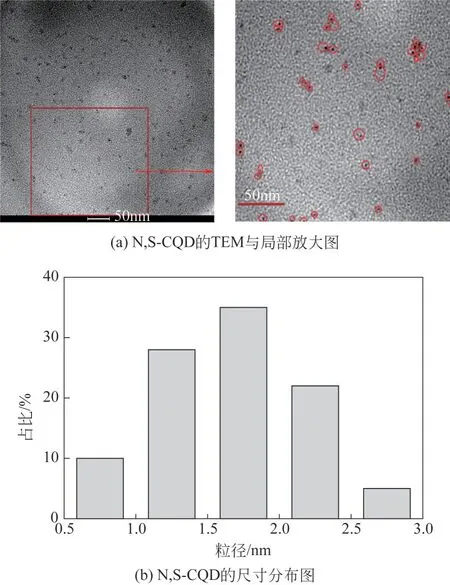
图4 N,S-CQD的TEM图及尺寸分布
采用FTIR 测试研究N,S-CQD 表面的官能团组成,结果如图5 所示。其中3444cm-1的宽峰归属于—NH与—OH的伸缩振动峰,2082cm-1处的吸收峰可能为芳环中C—H 的泛频峰,1637cm-1的吸收峰为C= = C的伸缩振动峰,680cm-1处的吸收峰是C—H 的特征峰。结果表明,N,S-CQD 周围存在羟基等含氧官能团,因此具有良好的水溶性[35]。

图5 N,S-CQD的FTIR谱图
对N,S-CQD 进行XPS 分析,探究其表面元素组成,如图6所示。将各元素原子百分数汇总到表2,从中可以发现主要存在C 和O 两种元素,其中C 元素相对含量为55.69%,O 元素相对含量为29.18%,说明N,S-CQD 表面含氧官能团数量非常多。除此之外,N,S-CQD 还存在一定量的N、S 及Cl 元素。由于本实验的电解液为氯化钠水溶液,而碳量子点的生成主要借助电解氧化体系中次氯酸根离子的氧化进攻,因此在N,S-CQD 中引入了Cl元素。
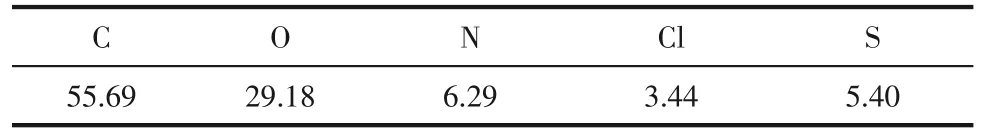
表2 N,S-CQD表面各元素相对含量 单位:%

图6 N,S-CQD的XPS全谱及各分峰谱图
为了更清晰地探究N,S-CQD 各元素的化学价态分布,分析N,S-CQD 的Cls、O1s、N1s 和S2p 高分辨谱图(图6),并将分峰拟合结果汇总到表3。发现N,S-CQD 中的C 元素有四种化学键合形式,分别为C= = C、C—N/C—S、C—O 以及C= = O,其中C= = C 和C—N/C—S 占比较高。O 元素有三种化学键合方式,峰面积占比从高到低依次为C—O、—OH、C= = O。N元素存在吡啶氮、氨基氮和吡咯氮三种形式。S元素以C—S和C—SO-x两种形式存在,162.44eV和163.6eV对应噻吩硫中的C—S共价键[13],168.7eV与169.8eV对应磺酸盐或硫酸盐中的C—SO-x(x=3、4)[36-37]。通过以上分析发现N,S-CQD 含有S元素,但FTIR图谱中未出现与S相关的吸收峰,这是由于S相关的FTIR吸收峰大多在400~800cm-1[38],且吸收峰强度较弱,而该处C—H 的特征峰非常明显,掩盖了与S相关的红外吸收峰。
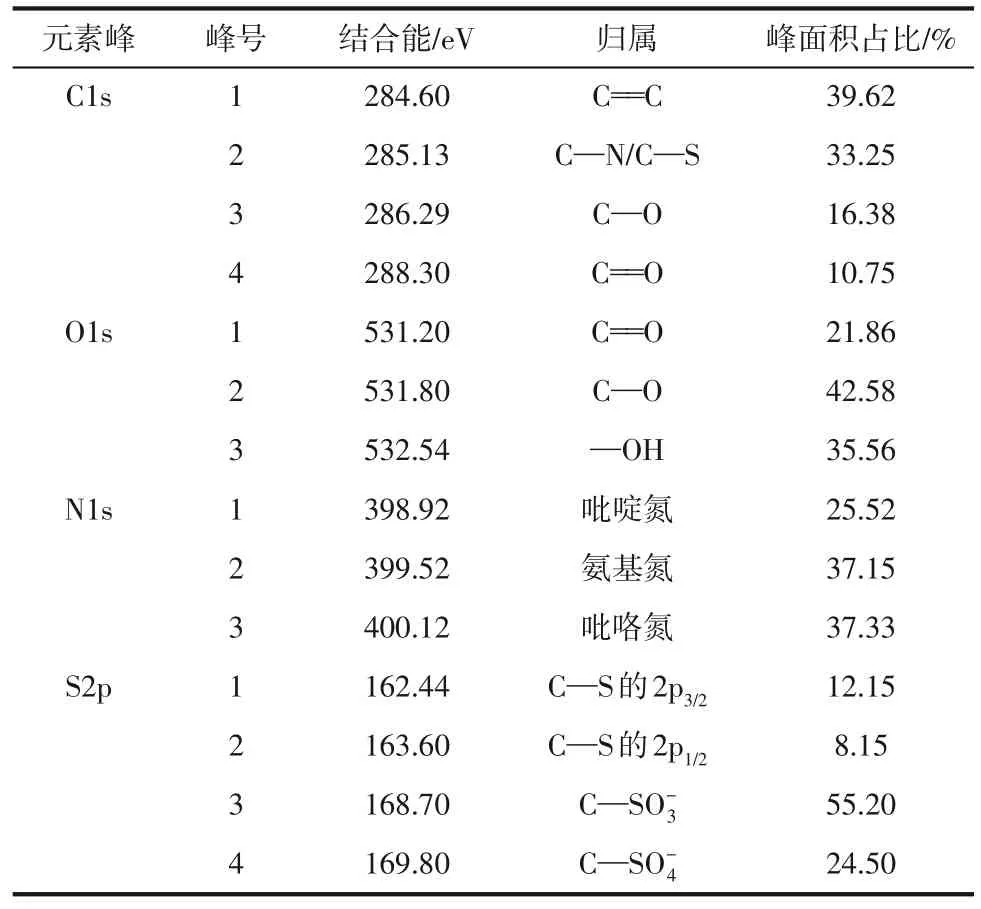
表3 N,S-CQD的C、O、N和S的吸收峰及其归属
2.4 N,S-CQD的光学性质分析
利用紫外可见分光光度计和荧光分光光度计研究碳量子点的光学性质。图7(a)展示了N,S-CQD的紫外吸收光谱,发现N,S-CQD 在可见光区吸收较弱,在紫外光区吸收明显,但未发现明显的特征峰,这是由于无定形炭的背景吸收过强,从而掩盖了一些化学键的特征吸收峰。图7(b)展示了N,S-CQD 的激发光谱和发射光谱,N,S-CQD 的最佳激发波长为280nm,最佳发射波长为313nm。

图7 N,S-CQD的紫外可见吸收光谱和荧光激发光谱、发射光谱
此外还考察了pH 对N,S-CQD 荧光强度的影响。如图8所示,N,S-CQD的荧光强度在中性环境中最高,在酸性环境中基本保持不变,这表明N,S-CQD在酸性和中性环境中具有良好的光稳定性,这可能源于异质元素掺杂对光稳定性的改善[37]。但N,S-CQD 荧光强度在碱性环境中随着pH 的增大而降低,这是由于羧基等含氧基团发生去质子化,影响与发光过程相竞争的非辐射跃迁过程,从而猝灭碳量子点荧光[39]。利用碳量子点在不同酸碱性环境中的荧光响应效应,可应用于生物传感器和生物成像领域[40]。
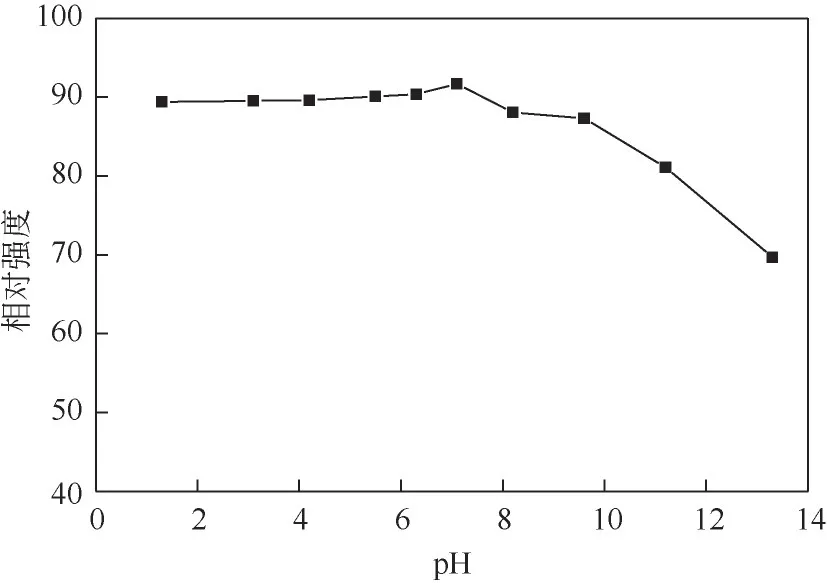
图8 不同pH下N,S-CQD的荧光强度
3 N,S-CQD荧光猝灭效应检测Fe3+
3.1 N,S-CQD检测Fe3+的选择性
如图9(a)所示,N,S-CQD 溶液在加入Fe3+后荧光猝灭,为了验证N,S-CQD 通过荧光猝灭效应检测痕量Fe3+的选择性,考察了常见的14种金属阳离子(Na+、K+、Ca2+、Zn2+、Mg2+、Cu2+、Fe3+、Ba2+、Al3+、Ni2+、Fe2+、Cr3+、Co2+、Ag+)对N,S-CQD 荧光强度的影响。如图9(b)所示,F0和F分别为空白和加入金属离子的N,S-CQD 溶液的荧光强度,发现不同金属离子在浓度为150µmol/L的条件下,只有Fe3+可以显著地降低N,S-CQD 的荧光强度,即N,S-CQD溶液对Fe3+的检测具有高选择性。这是因为相比其他金属离子,Fe3+对N,S-CQD 表面的N和O有更高的热力学亲和力和更快的螯合过程[39,41-42]。因此在排除其他离子干扰的条件下可以建立荧光猝灭检测Fe3+的方法。

图9 不同N,S-CQD体系的荧光光谱和加入不同金属离子后N,S-CQD的相对荧光强度
3.2 N,S-CQD检测Fe3+的灵敏性
基于N,S-CQD 对检测痕量Fe3+有良好的选择性,进一步考察了不同浓度Fe3+对N,S-CQD荧光强度的影响。结果如图10(a)所示,发现N,S-CQD的荧光强度随着Fe3+浓度的增大而逐渐降低。以N,S-CQD 溶液的相对荧光强度ΔF(ΔF=F0-F)为纵坐标,Fe3+的浓度为横坐标,进行线性拟合。如图10(b)所示,当Fe3+浓度在15~150µmol/L 范围内时,与ΔF有良好的线性关系,线性方程为ΔF=0.2697C+34.309。其中C为Fe3+的浓度,线性系数R2=0.9924,计算得最低检出限L=1.22µmol/L(其中S/N=3),优于之前报道中检测Fe3+得到的最低检出限[36,43](分别为4µmol/L 和2.2µmol/L),分析认为S元素的引入进一步增强了N,S-CQD中N元素的诱导效应,因而提高了检测方法的灵敏度[19]。上述分析表明当Fe3+浓度在15~150µmol/L 范围内时,N,S-CQD对痕量Fe3+的检测表现出良好的选择性和较高的灵敏度。
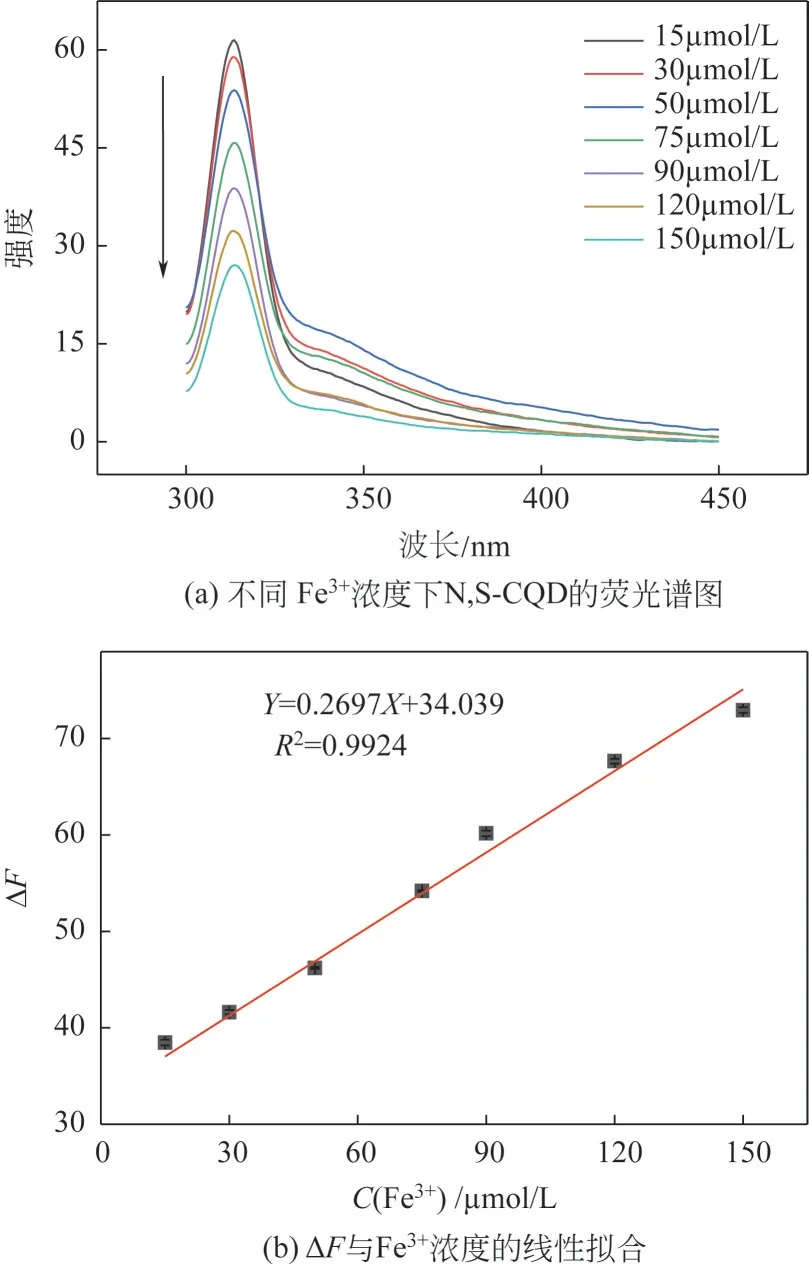
图10 不同Fe3+浓度下N,S-CQD的荧光谱图和ΔF与Fe3+浓度的线性拟合
3.3 N,S-CQD检测Fe3+的机理
为探究Fe3+对N,S-CQD荧光的猝灭机理,对N,S-CQD 的不同体系进行红外光谱测试。结果如图11(a)所示,在3444cm-1处的吸收峰为—OH和N—H的伸缩振动峰,1637cm-1处的吸收峰为C= = C 的伸缩振动峰,加入Fe3+之后两个吸收峰都发生红移,这是由于Fe3+会与分布在N,S-CQD 表面的—OH 与N—H 发生螯合作用[30]。这一点也可从紫外光谱中得到印证,如图11(b)所示,对比未加入Fe3+的N,S-CQD 溶液体系,发现加入Fe3+后在292nm处出现了一个吸收峰,表明Fe3+与—OH、N—H 发生了螯合作用。另外N、S 掺杂和碳量子点中大量的共轭sp2杂化轨道使得N,S-CQD 含有丰富的电子,在光照下,N,S-CQD中激发态电子会转移到Fe3+的半充满3d轨道上,导致非辐射电子-空穴的重新转移和复合,从而导致荧光猝灭现象[13,39]。
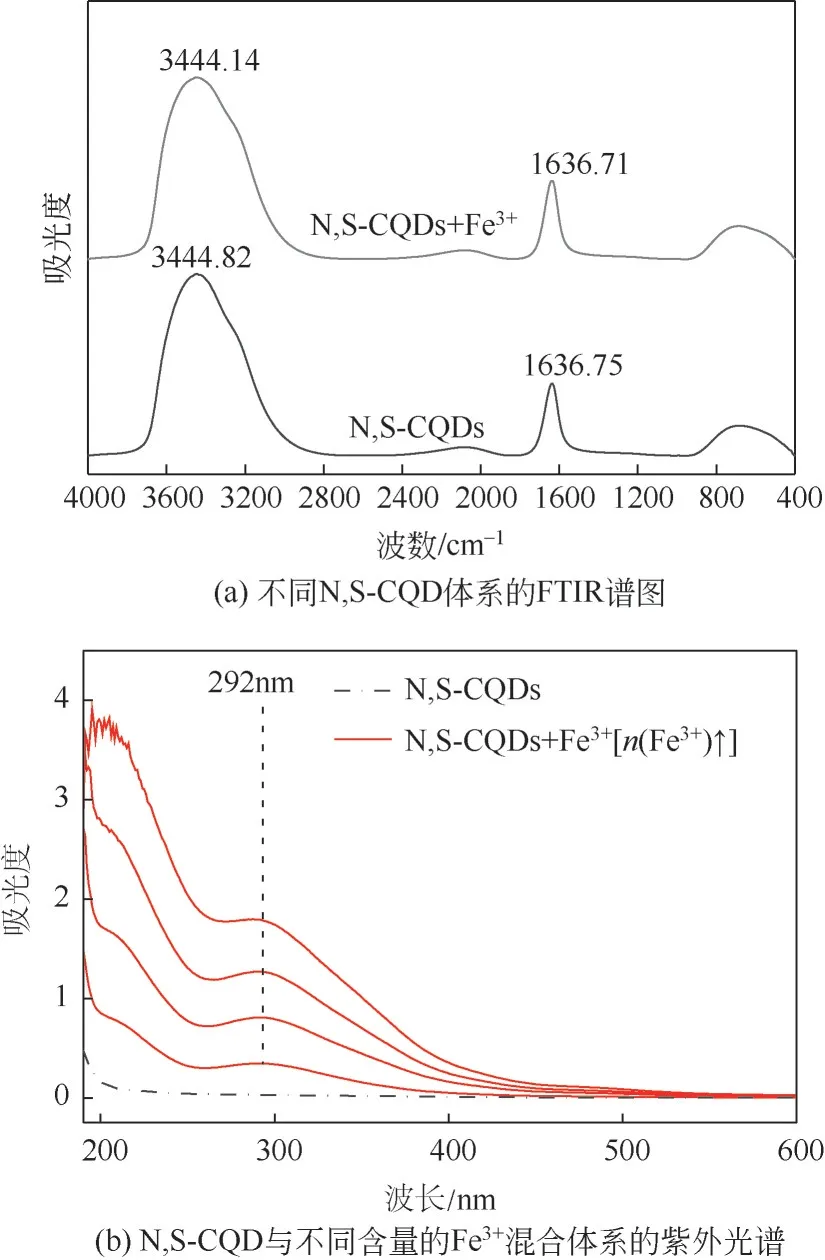
图11 不同N,S-CQD体系的FTIR谱图和N,S-CQD与不同含量的Fe3+混合体系的紫外光谱
4 结论
本文以褐煤为碳源,将褐煤与氯化钠溶液配制成水煤浆,对比氨水、乙二胺、硫脲和磷酸氢二铵作为掺杂剂的效果,以荧光量子产率为指标,选取掺杂效果最好的硫脲作为助剂,通过电解氧化法制备得N,S-CQD。N,S-CQD 的荧光量子产率为1.60%,最佳激发波长为280nm,最佳发射波长为313nm,且在酸性和中性环境中有着良好的光学稳定性。通过TEM、XPS 和FTIR 等表征手段分析发现,N,S-CQD 分散较均匀,平均粒径为1.66nm;N,S-CQD 表面存在丰富的—OH、—COOH 和C= = O等含氧官能团,并且含有一定量的N、S元素。以上结果说明以褐煤为原料通过电化学氧化法可以制备碳量子点,并且通过掺杂法能成功引入异质元素,优化了碳量子点的光学性质。最后将N,S-CQD用于检测Fe3+,通过实验发现N,S-CQD 对Fe3+有良好的选择性,当Fe3+浓度在15~150µmol/L 内时,Fe3+对碳量子点的荧光猝灭程度与Fe3+浓度成较好的线性关系,且最低检出限L=1.22µmol/L,对比发现检出限有所降低,表明N,S-CQD检测痕量Fe3+有着较高的灵敏度。并且探究了荧光猝灭机理,对比分析N,S-CQD加入Fe3+前后体系的荧光光谱、红外光谱和紫外光谱,发现Fe3+与N,S-CQD 表面的—OH 与N—H发生螯合作用使得荧光猝灭。
