Static and dynamic evolution of CO adsorption on γ-U (1 0 0) surface with different levels of Mo doping using DFT and AIMD calculations
2023-10-13JunWeiLiWeiMinJiaChongLiuShaShaLvJinTaoWangZhengCaoLi
Jun-Wei Li · Wei-Min Jia · Chong Liu · Sha-Sha Lv · Jin-Tao Wang · Zheng-Cao Li
Abstract Alloys of uranium and molybdenum are considered as the future of nuclear fuel and defense materials.However, surface corrosion is a fundamental problem in practical applications and storage.In this study, the static and dynamic evolution of carbon monoxide (CO) adsorption and dissociation on γ-U (1 0 0) surface with different Mo doping levels was investigated based on density functional theory and ab initio molecular dynamics.During the static calculation phase, parameters, such as adsorption energy, configuration, and Bader charge, were evaluated at all adsorption sites.Furthermore, the time-dependent behavior of CO molecule adsorption were investigated at the most favorable sites.The minimum energy paths for CO molecular dissociation and atom migration were investigated using the transition state search method.The results demonstrated that the CO on the uranium surface mainly manifests as chemical adsorption before dissociation of the CO molecule.The CO molecule exhibited a tendency to rotate and tilt upright adsorption.However, it is difficult for CO adsorption on the surface in one of the configurations with CO molecule in vertical direction but oxygen (O) is closer to the surface.Bader charge illustrates that the charge transfers from slab atoms to the 2π* antibonding orbital of CO molecule and particularly occurs in carbon (C) atoms.The time is less than 100 fs for the adsorptions that forms embryos with tilt upright in dynamics evolution.The density of states elucidates that the overlapping hybridization of C and O 2p orbitals is mainly formed via the d orbitals of uranium and molybdenum (Mo) atoms in the dissociation and re-adsorption of CO molecule.In conclusion, Mo doping of the surface can decelerate the adsorption and dissociation of CO molecules.A Mo-doped surface, created through ion injection, enhanced the resistance to uranium-induced surface corrosion.
Keywords Adsorption and dissociation · Uranium · CO molecule · Density functional theory · Ab initio molecular dynamics
1 Introduction
Uranium (U) holds the distinction of being the naturally occurring element with the highest atomic number (92) and relative atomic mass that has been discovered [1].It was found that uranium atoms release a large amount of energy through fission reactions.Uranium has been used in a wide range of industrial fields, such as nuclear power and aerospace [2–4].There are three lattice structures of uranium[3]: alpha-uranium (α-U) with orthorhombic structure, betauranium (β-U) with body-centered tetragonal, and gammauranium (γ-U) with body-centered cubic.The conversion temperatures from α to β, and β to γ are 941 K and 1047 K,respectively.γ-U lattice structures are stabilized in uranium alloys by adding alloying elements with similar crystal structure and atomic size, including molybdenum (Mo), zirconium (Zr), niobium (Nb), and titanium (Ti) [5–9].U–Mo alloys are widely used in the nuclear industry because of their excellent corrosion resistance and mechanical properties.Therefore, it is considered a promising nuclear fuels[7, 10, 11].
Surface corrosion is a typical problem owing to its special electronic structure and strong correlation with uranium 5felectrons in practical applications and the storage of uranium–molybdenum (U–Mo) alloys [12].The electrochemical properties of uranium are as active as those of aluminum, both of which are susceptible to corrosion.Uranium can react with oxygen (O2), hydrogen (H2), water(H2O), carbon dioxide (CO2), and carbon monoxide (CO)molecules at room temperature as per previous studies[13–15], and hydrogen embrittlement can easily induce material powdering and burning due to the reaction between uranium and hydrogen [16].Additionally, uranium oxide is formed after the reaction between uranium and hydrogen.The oxide layer peels offafter reaching a certain thickness,leading to further inward corrosion [17].Several researchers investigated the surface corrosion of uranium and uranium alloys.In the studies, reaction results were obtained with respect to small molecules interacting with material surfaces across various scales, from macro to micro, by utilizing techniques, such as Auger electron spectroscopy(AES) [18] and X-ray photoelectron spectroscopy (XPS)[19], among others [20].Microscopic mechanisms were explored through surface reactions.For instance, Tellurium(Te) and O belong to the same group on the periodic table.A study by Wang et al.[21] examined the effect of pure Nickel (Ni) on the corrosion behavior of a GH3535 alloy in a Te vapor environment.Their findings suggested that the GH3535 alloy can resist Te corrosion, but at the expense of pure Ni.However, the study of surface corrosion using experimental methods has certain limitations due to localized experimental conditions and the unique properties of uranium.
Recently, researchers have examined the theoretical mechanism of reactions between metals and small molecules.The interaction between hydrogen and 4dalloying atoms in Ni-based alloys has been studied using the density functional theory (DFT) [22].The larger size of the 4datom induces stronger extrusion in the supercell,resulting in a greater repulsive force on the H atom.For uranium, researchers intend to study small-molecule adsorption and dissociation on uranium and U alloys using a theoretical method, which is considered the initial stage of surface corrosion.Research has been conducted on the performance and mechanisms of adsorption, dissociation,and diffusion, particularly with molecules such as O2, H2,and H2O, on the surfaces of α-U, γ-U, Uranium-titanium(U-Ti), and U–Mo [23–27].The pronounced impact is attributed to the adsorption of carbon oxide on uranium and its alloys.
The adsorption of carbon oxide on a U surface has not been as extensively studied as that of hydrogen and oxygen,despite attracting substantial theoretical interest.Prior studies primarily focused on static adsorption behavior, which involved determining different adsorption site configurations, studying structural relaxation, exploring adsorption energy at various sites, identifying optimal adsorption sites,and analyzing related parameters such as charge transfer,work function, and density of states.However, the dynamic behavior of small-molecule adsorption, molecule dissociation, and atom migration remain under-studied in extant studies, making it challenging to ascertain a comprehensive mechanism for the initial stage of surface corrosion.Consequently, it is crucial to consider the evolution of adsorption and dissociation in further studies.
The evolution of the dynamics of CO adsorption and its dissociation on γ-U (1 0 0) surface with different Mo doping was investigated using density functional theory,ab initio molecular dynamics (AIMD), and climbing image-nudged elastic band method (CI-NEB) [28].This study is divided into four stages in this work: (i) Static calculations were performed on the adsorption of CO molecules in three reconstruction configurations for the Mo-doped U (1 0 0) surface, examining aspects, such as adsorption energy, configuration, bonding type, at all sites and optimal adsorption sites in different slabs.This foundational research is critical for subsequent analyses.(ii)The study separately elucidates the crystal orbital Hamilton population (COHP), charge transfer, and total density of states (TDOS) of CO molecules at the optimal sites.(iii)The evolution of the structure and two-dimensional differential charge density in the optimal adsorption models for CO adsorption is depicted using AIMD at 298.15 K.(iv) The minimum energy paths (MEPs) for CO molecule dissociation were explored.
2 Numerical simulation details
The Vienna ab initio simulation package (VASP 5.4.4) was used for DFT, AIMD, and CI-NEB calculations [29, 30].The projector-augmented wave (PAW) pseudopotential method is described in the electron–ion interaction section, suggesting an efficient plane-wave pseudopotential package in the electronic structure calculations of the surface model.The electron exchange-correlation is calculated by the Perdew-Burke-Ernzerhof (PBE) potential in Generalized Gradient Approximation (GGA) based on the Perdew–Burke–Ernzerhof exchange–correlation potential [31, 32].This effect was neglected for spin polarization [33].The plane-wave cut-offwas set to 500 eV.
The lattice parameter of γ-U is optimized using a bodycentered cubic (bcc) crystal cell, with an 11 × 11 × 11 Monkhorst–Pack k-point mesh employed for Brillouin zone integration.The lattice parameter is calculated to be 3.433 Å,which aligns well with the theoretical value of 3.532 Å[34].A slab comprised of five U atomic layers is created to simulate the γ-U (1 0 0) surface based on the computed lattice constant of γ-U.The dimensions of the simulation box are 6.8668 Å, 6.8668 Å, and 21.8668 Å for length, width, and height, respectively.Three Mo doping configurations were constructed, namely a single Mo atom replacing a U atom on the surface; a Mo atom substituting a U atom on the subsurface; and four Mo atoms replacing the entire top layer of U atoms on the surface.These reconstruction configurations were designated as Mo-U, Sec-Mo-U, and 4Mo-U for ease of subsequent analysis.
The bottom three layers were fixed and top two layers were relaxed in the slab calculation for structural optimization and AIMD simulations.K-point meshes of 5 × 5 × 1 and 7 × 7 × 1 were established for structural optimization and density of states, respectively.Additionally, the CO molecule was optimized and enclosed in a box with a side length of 10 Å.The computed bond length was found to be 1.144 Å, which closely aligns with the theoretical value of 1.129 Å.A vacuum region along thez-axis was chosen to isolate the periodic images, maintaining a thickness of 15 Å.Dipole correction was implemented to negate the dipole moment induced during CO adsorption [35].
Adsorption energy (Eads) is obtained from the following equation:
whereEslab+Adenotes the total energy after CO adsorption andEAdenotes the CO molecule energy.The negative adsorption energy demonstrates the stability of the adsorption system, and a larger absolute value of the adsorption energy indicates a more stable system.Conversely, a positive adsorption energy indicates that the system is unstable after adsorption.
AIMD calculations were conducted using the Verlet algorithm over 1000 fs with a timestep of 1 fs, based on the canonical ensemble (NVT ensemble).The initial velocities of the CO molecules were assigned in accordance with the Maxwell–Boltzmann distribution atT= 298.15 K.A Langevin thermostat [36, 37] was used to maintain the surrounding temperatures at 298.15 K, both at the start and end.The CI-NEB method was deployed to explore the diffusion of C and O atoms on the slab following CO adsorption.This method accurately determines the minimum energy paths and energy barrier, and also ensures the saddle point following convergence is the transition state.Energy convergence was established when the energy difference was less than 1 × 10–6eV for adjacent iteration steps, and force convergence was defined as a residual force per atom less than 2 × 10–2eV/Å.Particularly, CI-NEB calculations were repeated until the energy difference between adjacent iteration steps dropped below 1 × 10–7eV.
3 Results and discussion
3.1 Configuration of adsorption and adsorption energy
The adsorption of CO molecules on Mo-U, Sec-Mo-U, and 4Mo-U at highly symmetrical sites was investigated using the following calculations: The adsorption sites consist of top, hollow, and bridge sites.The adsorption sites on the Mo and U surface atoms must be considered under different chemical circumstances.The initial configuration comprised horizontal (Hor) and vertical (Ver) adsorption at each adsorption site.There are two adsorption categories for CO molecules, in which the O or C atoms are closer to the surface in the vertical direction.
The top and side views of adsorption models of CO molecule on Mo-U, Sec-Mo-U and 4Mo-U are shown in Figs.S1, S3 and S5, respectively, before structural relaxation.The top and side views of adsorption models of CO molecule on Mo-U, Sec-Mo-U and 4Mo-U are shown in Figs.S2, S4 and S6, respectively, after structural relaxation.The simplifications of the different adsorption configurations are described; for example, T-Hor, H-Hor, and B-Hor represent the top horizontal, hollow horizontal,and bridge horizontal, respectively.TMo-Hor and TU-Hor represent the top horizontal configurations of Mo and U atoms, respectively.Additionally, B-Ver and B-Ver-O represent the adsorption sites of the vertical bridge,where C and O atoms are closer to the surface, respectively.Furthermore, C and O atoms bonded to the slab surface atoms.The adsorption of CO molecules occurs via chemical adsorption, but CO does not dissociate after adsorption, apart from the HU-Hor and B-Hor configurations of Sec-Mo-U.This indicates that the CO molecule tends to rotate and tilt upright with stable adsorption and is approximately vertical to the surface in the final state.The C atom was close to the surface in the vertical direction.The phenomenon is also found in the CO molecule adsorption on α-U (0 0 1) surface [38].However, CO does not bond to slab atoms in the vertical adsorption configuration when the O atom is close to the surface.In some configurations, the CO molecule is far from the slab after structural relaxation.
Figure 1 shows a schematic of the CO single-molecule orbitals [39].The highest occupied molecular orbital(HOMO) and lowest unoccupied molecular orbital(LUMO) of free state CO molecule are the 5σ orbital and 2π* orbital, respectively, as shown in Fig.1.The 5σ orbital electrons were mainly concentrated near the C atomic side.The 2π* orbital is parallel to the C-O bond axis, which easily overlaps with thedorbitals of the U and Mo atoms on the slab.The CO molecule interacts with the substrate atoms mainly by bonding the C atom to the atoms, according to the matching requirement of the orbital orientation.Therefore, it can be inferred that the CO molecule is vertically adsorbed onto the surface in most configurations.
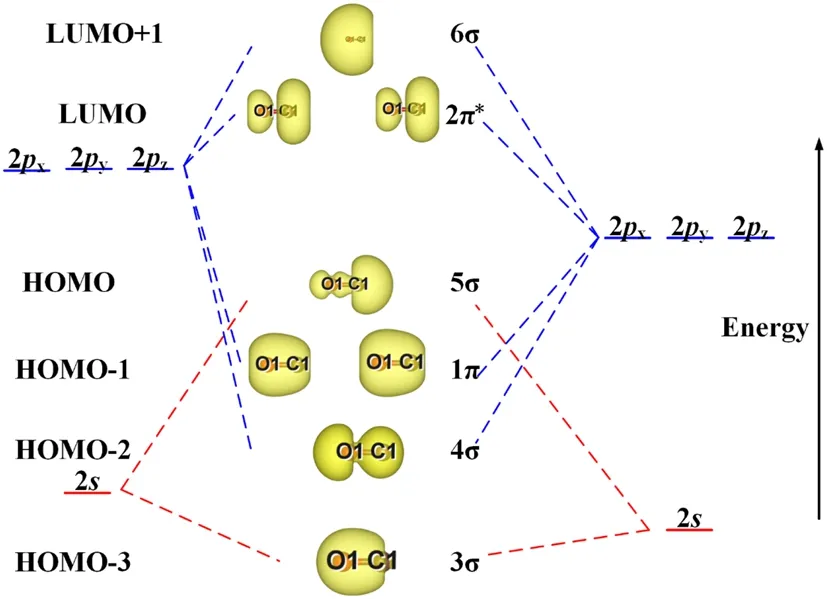
Fig.1 (Color online) Schematic diagram of CO molecular orbitals
Figure 2 shows the top and side views of the optimal adsorption configurations of CO molecules on the slab after structural relaxation.H-Hor (hollow horizontal), B-Hor(bridge horizontal), and H-Ver (hollow vertical) were the optimal adsorption configurations for Mo-U, Sec-Mo-U, and 4Mo-U, respectively.The adsorption of CO molecules on slabs involves chemical adsorption, which is followed by bonding to the surface atoms.The adsorbed CO molecules tended to tilt upright on the surface in the optimal configurations of Mo-U and 4Mo-U.The CO molecule completely dissociates in the optimal configuration of Sec-Mo-U, and C atoms are bonded not only to surface atoms but also to Mo atoms on the subsurface.
Tables S1, S2, S3 show the adsorption energies and geometrical parameters of CO molecules on Mo-U, Sec-Mo-U,and 4Mo-U, respectively.The chemical adsorption of the CO molecules is shown in Tables S1, S2, S3.This indicates thatdC-Oincreases when compared with the bond length of the CO-free state after adsorption, leading to the deterioration of the C-O bond strength.
Table 1 lists the adsorption energy and geometrical parameters of the optimal configuration of CO molecules on the slab.The absolute value of the adsorption energy of Sec-Mo-U was larger than those of Mo-U and 4Mo-U,indicating that dissociated adsorption was more stable than undissociated adsorption.The absolute value of the adsorption energy of Mo-U was larger than that of 4Mo-U, indicating a decrease in Mo surface doping, leading to more stable CO adsorption.It is determined that thedBondbetween different bonding atoms in Table 1 is close to the bond length of U-C in UC (2.480 Å) [40], Mo-C in Mo2C (2.112 Å)[41], and O-U in UO2(2.370 Å) [42], indicating that a stable chemical bond is formed between C or O atoms and U or Mo atoms on the slab.
Figure 3 depicts the COHP of the C and O atoms on the surface atoms for the optimal adsorption configurations of Mo-U, Sec-Mo-U, and 4Mo-U.The LOBSTER program[43, 44] was used to calculate the COHP for the interatomic interactions.Positive and negative COHP results signify bonding and antibonding interactions, respectively.In Fig.3, the COHP of the C or O atoms is positive for the bonding atoms of the slab below the Fermi level,indicating bonding interactions between the CO molecule and slab atoms following CO adsorption.The strength of the interaction between two atoms is directly determined by the Integrated Crystal Orbital Hamilton Population(ICOHP)) [45], which is calculated using different energies below the Fermi level.An increase in bonding strength is associated with an increased ICOHP.Tables S3 and S4 display the ICOHP values for the COHP.As Table S4 shows, the ICOHP values for the CO molecule bonding to slab atoms follow a descending order of Sec-Mo-U, Mo-U, and 4Mo-U.This trend verifies the variation in adsorption stability of the optimal configuration in different slab models according to the ICOHP results.Additionally, Table S4 lists the ICOHP of C–O for the free state and optimal adsorption configurations.The adsorption of CO molecules results in a reduction of the ICOHP of C-O.It was also observed that a higher interaction intensity between the C or O atoms and the slab atoms correlates with a larger degree of ICOHP degradation.

Fig.2 (Color online) Top and side views of the optimal adsorption configurations of CO molecules on the slab after structural relaxation: (a) Mo-U;(b) Sec-Mo-U; (c) 4Mo-U
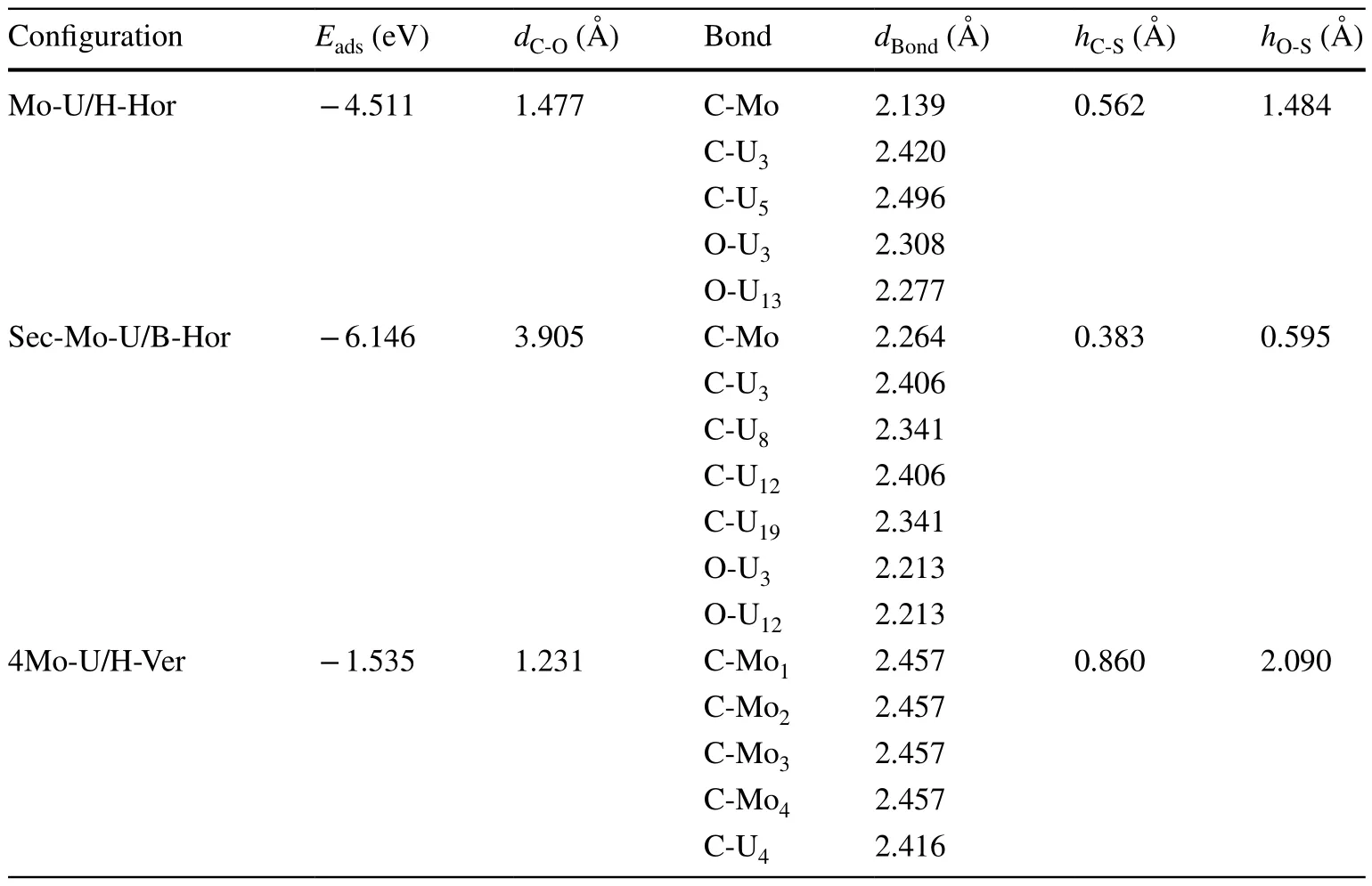
Table 1 Adsorption energy and geometrical parameters in the optimal adsorption configurations of CO molecule adsorption on the slab: Eads(the adsorption energy), dC-O(the distance between C and O atom), Bond (the C and O atom bonding with the slab surface atom), dBond (bond length), and hC-S/hO-S (the distance between the C/O atom and slab surface after adsorption)
Table S5, S6, S7 shows the net charge distribution numbers for CO adsorption on Mo-U, Sec-Mo-U, and 4Mo-U for all adsorption configurations.The net charge is expressed as follows [46]:
whereqatom,qBader, andqvalencedenote the net atomic, Bader,and valence electrons, respectively.This indicated that the net charge of the CO molecule increased after adsorption,according to the charge-transfer results.Simultaneously,the slab lost electrons according to the principle of charge conservation, demonstrating that the electrons were mainly transferred from the slab atoms to the CO molecule.There are differences in the layers for the change in electrons after adsorption, causing the loss of the net charge number to mainly occur in the top two layers, especially in the first layer.It is believed that the C or O atoms mostly interact and transfer charge with the U and Mo atoms at the surface and subsurface.
Table 2 lists the net charge distribution numbers for the optimal configurations of CO adsorption on the slab.The sum net charge number ranges from 0.9178 to 2.7506 e for C and O atoms in the optimal models.The accumulation of charge is found to be closer to or larger than 1 e, indicating that the CO molecules form stable bonds with the slab atoms.The sum net charge number for the CO molecules exhibits a descending order with Sec-Mo-U, Mo-U, and 4Mo-U, which directly illustrates the interaction stability of the CO molecules on different slabs.Furthermore, the net charge gained by the C atoms is significantly larger than that by the O atoms, indicating that electrons are mainly transferred through the C atoms from the slab to the CO molecule.
Figure 4 displays the TDOS of the slabs both before and after CO adsorption in the most stable configuration.Upon CO adsorption on slabs, new peaks locally form within the ranges of - 25 to - 20 eV and - 10 to - 5 eV.Figure 5 illustrates the TDOS of the CO molecule before and after its adsorption on the slab.It is observed that in the energy range of –5 to 5 eV, the HOMO and LUMO of the CO molecule exhibit significant interaction with slab atoms during CO molecule adsorption.The heights of the HOMO and LUMO orbitals undergo a substantial decrease, indicative of orbital delocalization.The Fermi level is elevated to a higher energy level, and the LUMO orbitals are filled with electrons due to electron transfer from the slab to the CO molecule.

Fig.3 (Color online) COHP of C and O atoms to the surface atoms which bond to CO molecule in the optimal adsorption configurations: (a) Mo-U; (b) Sec-Mo-U; (c) 4Mo-U
The integral value of the electron state in Fig.5c is larger than those of (a) and (b) at energy levels ranging from - 5 to 5 eV, leading to more nonlocalized electrons in the adsorption.The LUMO of CO molecule is 2π* orbital,which is comprised of thepatomic orbital for C and O.It can be determined that the energy level position of 2π*orbital is closer to the C atomic orbital than that of O atomic in Fig.1.Therefore, the injecting electrons of 2π* antibonding orbital are mainly concentrated on the surrounding of C atoms, illustrating that the gaining of C atoms is more than that of O atoms in CO adsorption.
3.2 Structural evolution of the optimal configurations
Figures 6, 7, 8 present the structural evolution from top and side perspectives, as well as the two-dimensional differential charge density [47] in the optimal configurations of CO adsorption on Mo-U, Sec-Mo-U, and 4Mo-U, using AIMD simulation at 298.15 K.Concurrently, Figures S7,S8, S9 illustrate the corresponding three-dimensional differential charge density evolution at 298.15 K.CO progressively approaches the surface and uprightly rotates and tilts on Mo-U at 63 fs, resulting in a vertical adsorption stage of the CO molecule, where the C atom is closer to the surface.An approximate adsorption stage for 4Mo-U is observed at 83 fs, as shown in Fig.8.The CO molecule and slab atoms are, respectively, enveloped by red and blue regions,resulting in substantial electron accumulation and depletion.Charge accumulation is observed at C and O atoms enveloped by red regions.Conversely, charge depletion occurs at the slab surface atoms adjacent to the C and O atoms, enveloped by blue regions.Additionally, at 0 fs in Figs.6a, 7, 8a,the interaction between the CO molecule and slab atoms is relatively weak.However, significant electron accumulation and depletion between the CO molecule and adjacent slab atoms can be observed as reaction time increases.After adsorption on Sec-Mo-U, the CO molecule is completely dissociated, as depicted in Fig.7, leading to the formation of bonds between the C and O atoms and the slab atoms.The spacing between C and O is 2.167 Å at 48 fs, which suggests a break in the C-O bond, indicating a transition state between the molecular state and the dissociation of the CO molecule.The spacings between the C and O atoms further expand to 3.620 Å and 4.239 Å at 128 fs and 345 fs,respectively.The O and C atoms are eventually adsorbed at the hollow sites of the slab, in light of the rearrangement of the slab surface atoms.The differential charge densities of the C and O atom accumulation regions are higher than those of Mo-U and 4Mo-U in the final state, suggesting that a larger charge transfer enhances the stability of the C-U,O-U, and C-Mo bonds.
Figure 9 presents the partial density of states (PDOS) at various evolution stages in the optimal configurations for CO adsorption at 298.15 K for (a) Mo-U, (b) Sec-Mo-U, and(c) 4Mo-U.The PDOS of the U and Mo atoms representsthe PDOS of the surface and subsurface atoms bonded to C or O atoms in the slab, as depicted in Fig.9.The PDOS of U and Mo atoms extend beyond the Fermi level, indicating the strong metallic nature of U and Mo atoms.The PDOS hybridization occurs between C and O atoms, resulting in a distinct peak in the energy range from - 12 to - 4 eV at 0 fs.The PDOS of the C and O atom peaks diminish, and the peak broadens and overlaps with the U and Mo atoms.The peak values decrease following the broadening of the peaks for the PDOS of C and O atoms caused by the adsorption of CO molecules.The hybridization with U and Mo atoms highlights the bonding characteristics.Additionally, as shown in Fig.9a, c, no C-O bond breakage occurs after CO adsorption, followed by severe overlapping hybridization observable between the C and O atoms on Mo-U and 4Mo-U from the initial to the final state.However, at 48 fs, there is no evident hybridization of the PDOS between the C and O atoms, implying a weakening interaction between the C and O atoms.Eventually, after 122 fs, the hybridization completely vanishes between the C and O atoms, signifying a lack of interaction between C and O atoms.Independent peaks of the PDOS of the C and O atoms hybridize with the U and Mo atoms, respectively.
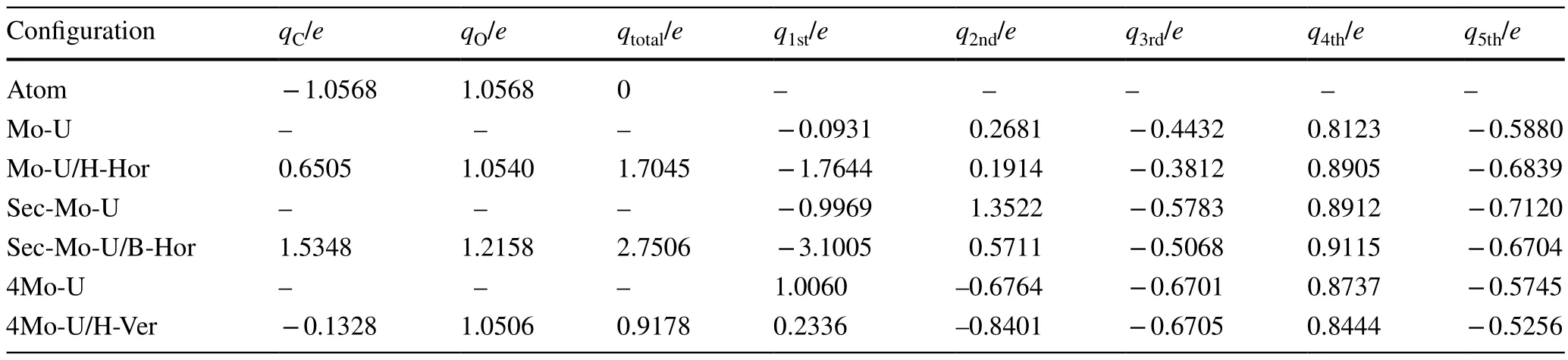
Table 2 Net charge distribution number in the optimal configurations of CO adsorption on the slab: qC and qO denote net charge number of C and O atoms, qtotal represents the sum net charge number of C and O atoms, q1st, q2nd, q3rd, q4th, and q5th represent the total net charge number of first to fifth layers on the slab, respectively
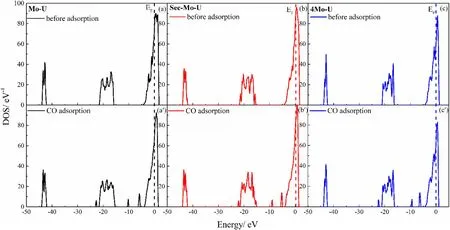
Fig.4 (Color online) TDOS of slabs before and after CO adsorption: before adsorption (a) Mo-U; (b) Sec-Mo-U; (c) 4Mo-U; after CO adsorption (a′) Mo-U; (b′) Sec-Mo-U; and (c′) 4Mo-U
3.3 Dissociation of CO molecule

Fig.5 (Color online) TDOS of CO molecule before and after adsorption on the slab: (a) before adsorption; (b) Mo-U; (c) Sec-Mo-U; (d)4Mo-U
The minimum energy paths were calculated for CO molecule dissociation in the optimal configurations of Mo-U and 4Mo-U using the CI-NEB method, which provided a more profound understanding of the further dissociation mechanism of CO molecules on the U–Mo slab after adsorption.MEP values were calculated for the optimal configurations of Mo-U and 4Mo-U.Studies on the dissociation process of Sec-Mo-U are not discussed in this section because of the dissociation of CO molecule into C and O atoms on Sec-Mo-U in the optimal configuration.Transient calculations were based on the steepest descent method.Simultaneously,a specified number of images were linearly interpolated between the initial and final configurations.More importantly, spring forces were added to the constrained optimization of the images.
Figures 10 and 11 show the energy profiles for the dissociation of the CO molecule and the diffusion of atoms on Mo-U and 4Mo-U, respectively, in conjunction with the PDOS for the CO molecule and bonded atoms on Mo-U and 4Mo-U in various states.A potential barrier of 0.074 eV exists when the O atom overcomes Mo-U, leading to the rupture of the C-O bond.The potential barrier increases to 1.411 eV when the O atom surpasses the 4Mo-U.The O atoms migrate to adjacent sites, resulting in the bonding of the O atoms and slab surface atoms on Mo-Uand 4Mo-U in the final state.It is also noteworthy that the energy needed to break the C-O bond of the CO molecule adsorbed on Mo-U is less than that on 4Mo-U.This can be attributed to the weaker interaction of the C-O bond on Mo-U than on 4Mo-U after adsorption, as reflected by the ICOHP values for the C and O atoms on Mo-U and 4Mo-U in Table S3.

Fig.6 (Color online) Evolution of structure with top and side views, and two-dimensional differential charge density in the optimal configuration of CO adsorption on Mo-U at 298.15 K.Red and blue color represent electrons accumulation and depletion, respectively

Fig.7 (Color online) Evolution of structure with top and side views, and two-dimensional differential charge density in the optimal configuration of CO adsorption on Sec-Mo-U at 298.15 K.Red and blue color represent electrons accumulation and depletion, respectively
The C 2pand O 2porbitals hybridize with U 6p, U 6d, U 5f, Mo 4p, and Mo 4dorbitals in the region of –5 eV in initial state, as shown in Figs.10b and 11b.However, this peak was split, and its shape broadened after a decrease in the peak value.This suggested the formation of new chemical bonds in the transition state.There was a severe decrease in the PDOS overlapping hybridization of the C and O orbitals,implying that the interaction of C and O atoms became weak in the transient and final states.There was no obvious overlapping hybridization for the PDOS of C and O atoms in the final state, illustrating that the CO molecule was completely dissociated on Mo-U and 4Mo-U.Meanwhile, the PDOS of C and O atoms overlapping hybridize with U and Mo atoms separately, according to the overlapping of density of states in black or red dotted bordered rectangle in the ‘Final state’in Figs.10b and 11b.Overlapping hybridization is mainly caused by the 2porbitals of C and O atoms anddorbitals of U and Mo atoms during the dissociation and re-adsorption of CO molecules.
Figure S10 shows the energy of the system and maximum force of each atom versus the number of iteration steps in the transition state calculation.This was terminated after 150 and 76 steps for the iterative calculations of Mo-U and 4Mo-U, respectively.The maximum force of each atom generally decreased with an increasing number of ionic steps.The energy of the systems gradually stabilized with an increase in the number of ionic steps for Mo-U and 4Mo-U.Both the force and energy reached the preset convergence standard,indicating that the iteration calculations were accurate for the CO molecule dissociation transition state on Mo-U and 4Mo-U.
4 Conclusion
The static and dynamic behaviors of CO adsorption on γ-U (1 0 0) surface at different levels of Mo doping were explored using DFT and AIMD.The adsorption of CO molecules on the slabs is predominantly chemical, except for the vertical orientation of the CO molecule.However, O atom is closer to the surface in the initial configuration.On theseslabs, the adsorbed CO molecules do not tend to dissociate, but the CO molecules do dissociate in the HU-Hor and B-Hor configurations of Sec-Mo-U.Moreover, the optimal adsorption configurations were hollow horizontal, horizontal bridge, and hollow vertical, with adsorption energies of - 4.511, - 6.146, and - 1.535 eV for Mo-U, Sec-Mo-U,and 4Mo-U, respectively.Bader charge analysis reveals that the charge transfer from surface atoms to the C atom exceeds that to the O atom, as theporbital of the C atom is closer to the 2π* orbital than the p orbital of the O atom.When the time was less than 100 fs, the CO molecule on the slab demonstrated a nascent form of adsorption, leading to the vertical adsorption of CO molecules, where the C atom was closer to the surface.At 48 fs, in the B-Hor configuration of Sec-Mo-U, the spacing between C and O is 2.167 Å, leading to the breakage of the C-O bond and dissociation of the CO molecule.
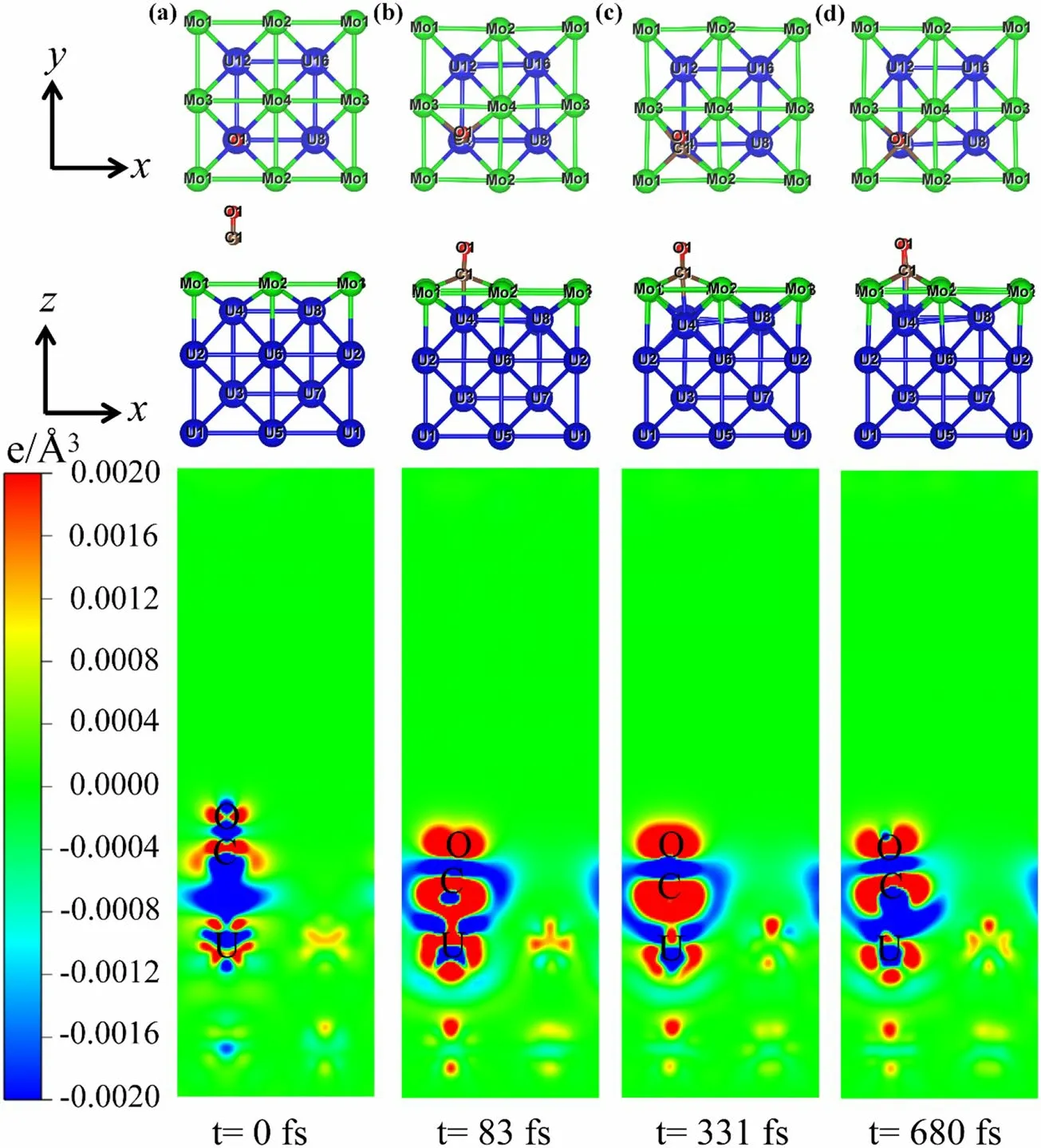
Fig.8 (Color online) Evolution of structure with top and side views, and two-dimensional differential charge density in the optimal configuration of CO adsorption on 4Mo-U at 298.15 K.Red and blue color represent electrons accumulation and depletion, respectively
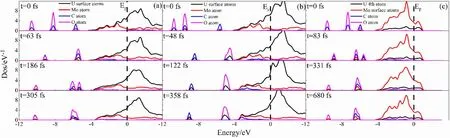
Fig.9 (Color online) PDOS of different evolution stages in the optimal configurations of CO adsorption at 298.15 K: (a) Mo-U; (b) Sec-Mo-U;and (c) 4Mo-U

Fig.11 (Color online) (a) Energy profile for CO molecule dissociation and atom diffusion on 4Mo-U (b) PDOS for CO molecule and bonding atoms on 4Mo-U at different states
There are two potential barriers of 0.074 eV and 1.411 eV encountered when the O atom surpasses Mo-U and 4Mo-U, respectively, leading to the breakage of the C–O bond.The O atoms migrate to the top of the Mo subsurface atoms and form a bridge between two adjacent Mo atoms on both Mo-U and 4Mo-U.Throughout the dissociation and re-adsorption of CO molecules, the 2porbitals of C and O atoms predominantly overlap with thedorbitals of U and Mo atoms.An increase in surface Mo doping not only elevates the difficulty of CO molecule adsorption but also raises the energy potential barrier of atom diffusion, thereby hindering the formation of an oxidation layer caused by CO adsorption on the uranium surface.This suggests that modulating Mo doping on the uranium surface can diminish the adsorption and dissociation of small molecules and enhance corrosion resistance during practical uranium storage and application.
Supplementary InformationThe online version contains supplementary material available at https:// doi.org/ 10.1007/ s41365- 023- 01287-z.
Author contributionsAll authors contributed to the study conception and design.Material preparation, data collection and analysis were performed by Wei-Min Jia, Chong Liu, Sha-Sha Lv, Jin-Tao Wang and Zheng-Cao Li.The first draft of the manuscript was written by Jun-Wei Li and all authors commented on previous versions of the manuscript.All authors read and approved the final manuscript.
FundingThis work was supported by the National Natural Science Foundation of China (Nos.11975135 and 12005017), the National Basic Research Program of China (No.2020YFB1901800).
Data availabilityThe data that support the findings of this study are openly available in Science Data Bank at https:// www.doi.org/ 10.57760/ scien cedb.j00186.00163 and https:// cstr.cn/ 31253.11.scien cedb.j00186.00163.
Declarations
Conflict of interestThe authors declare that they have no competing interests.
杂志排行

Nuclear Science and Techniques的其它文章
- Calculation of microscopic nuclear level densities based on covariant density functional theory
- Employing adaptive fuzzy computing for RCP intelligent control and fault diagnosis
- Comparison between 4D robust optimization methods for carbon-ion treatment planning
- Aperture shape optimization in intensity-modulated radiation therapy planning
- Discrimination of pp solar neutrinos and 14C double pile-up events in a large-scale LS detector
- Dynamic scaling characteristics of single-phase natural circulation based on different strain transformations