SMARCAL1基因新发突变相关的Schimke免疫-骨发育不良1例报告
2023-10-13张蝶,吕玲
张 蝶,吕 玲
(天津市儿童医院、天津大学儿童医院内分泌科; 天津市儿科研究所、天津市儿童出生缺陷防治重点实验室,天津 300074)
Schimke免疫-骨发育不良(SIOD)是由SCHIMKE等[1]于1974年报道,是一种常染色体隐性遗传的罕见疾病,由SMARCAL1基因突变导致[2],国外既往报道的发病率为1/(300~100)万,国内目前发病率不详,但已有明确诊断的病例报道[3-5]。Schimke免疫-骨发育不良是一种进行性加重且可累及多个系统的疾病,主要的临床表现为短躯干型身材矮小(躯干短、腰椎前突、腹部膨隆)、特殊面容(塌鼻梁、短颈)、皮肤色素沉着、肾病综合征,患儿因脊柱骨骺发育不良,影像学检查常存在椎体扁平,几乎所有患儿均有肾脏损害,且呈进行性加重,早期表现为蛋白尿,肾脏活检病理类型以局灶节段性肾小球硬化多见[6-7],另外此疾病常合并T细胞免疫缺陷、内分泌腺功能异常、脑发育受损等。本文收集1例SIOD患儿的临床资料,并报告1处新发SMARCAL1基因变异位点。
1 临床资料
1.1 一般情况
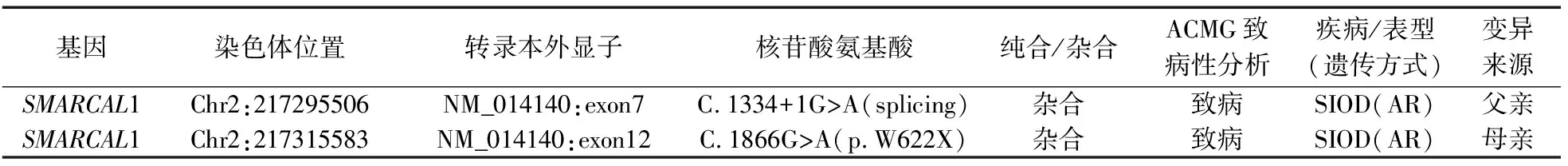
患儿,男,就诊年龄为5岁3个月,因“发现生长落后5年余伴胸廓畸形2年”于2022年3月21日入院。入院查体:身高82.2 cm(<-3SD),体重11.1 kg,头围46.5 cm,胸围49 cm,腹围44 cm,指尖距85.5 cm,坐高43.2 cm,坐高/下身长1.1:1( 血常规:血红蛋白139 g·L-1,白细胞5.22×109L-1,中性粒细胞比例56.3%,淋巴细胞比例31.2%,血小板292×109L-1。尿常规:尿比重(SG)1.024,PH 5.5,蛋白3+,镜检红细胞0~2个·HP-1,白细胞0~1个·HP-1。粪便常规未见异常。生化检查:钠140 mmol·L-1,钾4.99 mmol·L-1,钙2.13 mmol·L-1,磷1.9 mmol·L-1,镁0.79 mmol·L-1,碱性磷酸酶176 U·L-1,谷丙转氨酶1 U·L-1,谷草转氨酶35 U·L-1,γ谷氨酰转肽酶7 U·L-1,总蛋白51.8(参考值61.0~79.0)g·L-1,白蛋白27.8(参考值39.0~54.0)g·L-1,球蛋白24(参考值15.0~34.0)g·L-1,肌酐24 μmol·L-1,尿素氮3.57 mmol·L-1;血脂:甘油三酯1.20 mmol·L-1(参考值0.00~2.26)mmol·L-1,胆固醇5.82(参考值0.00~5.20)mmol·L-1,高密度脂蛋白1.96(参考值>1.45)mmol·L-1,低密度脂蛋白3.51(参考值0.00~2.59)mmol·L-1。甲功三项:T3 1.03 nmol·L-1,T4 130.04 nmol·L-1,TSH 1.59 mU·L-1。ACTH+皮质醇:ACTH 29.56 pg·mL-1,皮质醇371.0 nmol·L-1。IGF-1 82(参考值22~208)ng·mL-1,IGF-BP 34.74(参考值1.1~5.2)μg·mL-1。甲状旁腺激素2.75(参考值1.6~6.9)pmol·L-1。25羟维生素D23.7 ng·mL-1,25羟维生素D38.5 ng·mL-1,D2+D314.20 ng·mL-1。24 h尿蛋白定量:1769.3 mg·24 h-1(159.4 mg·kg-1·d-1)。 患儿影像学表现见图1。 骨龄:相当于3岁6个月(图1A)。左上肢长骨像:左肱骨、左尺桡骨、左肘关节诸骨骨质未见异常。脊柱全长侧位片:颈椎椎体变扁,椎间隙变窄,部分胸椎及腰椎椎体呈前高后低改变,脊柱生理曲度存在,部分椎间隙变窄(图1B)。头+垂体MRI:双额顶叶白质区多发点线样长T1长T2信号影,考虑血管周围间隙增宽;右侧小脑半球线样及簇状低信号影,考虑发育性静脉异常(图1C),垂体形态变扁,高度2 mm。 本例患儿以矮小就诊,临床表现存在短躯干型身材矮小、特殊面容、全身散在色素沉着,实验室检查结果示患儿存在低蛋白血症、高胆固醇血症、蛋白尿,即提示患儿存在肾病综合征,影像学检查提示本患儿存在脊柱干骺端发育异常,综合上述情况,考虑本例患儿为综合征相关矮身材,故对其进行相关基因学检查。 征得患儿父母同意后,提取患儿及患儿父母外周血2 mL,提取基因组DNA,提取之后进行质检,质检合格后将1~3 μg的基因组DNA片段化至150 bp平均大小,然后进行末端修复、接头连接和PCR扩增,进一步测序,测序后,将原始数据进行生物信息学分析并与多个数据库关联,并进行变异有害性预测。 基因变异结果:检测到SMACAL1基因变异,分别为C.1334+1G>A(导致氨基酸改变splicing的剪接突变,为曾经报道过的致病性变异[4-6])和C.1866G>A(导致氨基酸改变P.W622X的错义突变,为既往未报道的变异)。C.1334+1G>A突变位点来源为患儿父亲,患儿父亲该位点为杂合变异,患儿母亲该位点无变异;C.1866G>A突变位点来源为患儿母亲,患儿母亲该位点为杂合变异,患儿父亲该位点无变异见表1、图2—3。 表1 患儿及其父母的基因变异结果 图2 来源于患儿父亲的基因变异图谱 图3 来源于患儿母亲的基因变异图谱 基因变异致病性分析:1)c.1334+1G>A(exon7,NM_014140),导致氨基酸改变splicing,为剪接突变。根据ACMG指南[8],该变异初步判定为致病性变异。2)c.1866G>A(exon12,NM_014140),导致氨基酸改变p.W622X,为无义突变。根据ACMG指南[8],该变异初步判定为致病性变异。 SIOD为常染色体隐性遗传性疾病,其诊断基于基因学检测结果及临床症状。SMACAL1基因变异是导致SIOD主要原因,以杂合复合变异常见[9-10],且即使具有相同位点的基因突变,临床表现可不同[11]。多数患儿以肾脏疾病就诊,表现为进行性加重的肾病综合征,且对激素治疗无效,预后较差;多数患儿死于感染及肾功能衰竭,但目前已有病例报道通过序贯干细胞肾移植后患儿肾功能恢复正常[12]。SIOD临床表现常存在身材矮小,典型表现为躯干短小,主要由于脊柱骨骺端发育不良导致[13-14],常伴有特殊面容、皮肤色素沉着、免疫缺陷、脑血管疾病等情况,另有报道[15]发现SIOD有合并脑电图异常的情况。既往病例报告[16]报道的患儿脑血管疾病考虑与肾脏疾病及免疫功能障碍导致血管闭塞性疾病及动脉粥样硬化相关,而本例患儿出现脑血管发育异常为既往报道的病例未报告的临床表现,目前与SIOD的相关性尚不明确。经检测,本例患儿存在SMACAL1基因变异,分别为C.1334+1G>A和C.1866G>A,且C.1866G>A的错义突变为既往未报道过的变异,结合本例患儿的临床症状,最终明确诊断为SIOD。 以“矮小”就诊的患儿,如合并肾功能损害、骨骺发育异常、特殊面容、皮肤色素沉着等临床表现,需警惕SIOD可能,明确诊断需基于基因学检测结果及临床症状。1.2 实验室检查
1.3 影像学检查
1.4 基因检测


2 讨论
