模板法合成平面大环四齿膦配体17-aneP4钼氧配合物
2023-10-12司梦月,廖骞
司 梦 月, 廖 骞
(大连理工大学 化学学院,辽宁 大连 116024 )
0 引 言


至今尚未有通用的合成大环膦配体的方法,且没有以第六副族过渡金属为模板合成平面大环四齿膦配体[9]的报道.平面大环四齿膦配体的合成大多以Cu(Ⅰ)、Ni(Ⅱ)、Pd(Ⅱ)或Pt(Ⅱ)为模板[10-16].模板法合成的大环膦配体很难甚至不可能从金属上脱除,因此大环四齿膦配体的配位化学也尚未得到深入研究[17].1994年报道的十元大环四齿膦配体的尺寸太小,不能完全包围中心金属Mo.配体扭曲成船型结构,交替的膦基团螯合到单个Mo中心.配体可以通过其他两个膦基团与第二个Mo中心配位,产生双金属配合物[18].
相比于氧杂环、氮杂环和硫杂环系统,大环膦配体的研究较少[19].这可能与磷原子的立体化学性质有关.与叔胺不同,膦的翻转势垒很高(125~146 kJ/mol),在室温下手性膦分子不会发生翻转.这意味着具有不对称膦基团的大环膦(PR1R2R3)是手性的,而大环膦配体通常含有3个及3个以上的磷原子,导致每个大环有多种可能的立体异构体[17,20].例如Pd(Ⅱ)的双齿膦配合物在乙醇中与两当量乙酰丙酮反应形成的大环膦配合物具有两种异构体,它们分别衍生自起始化合物的(RSSR)-形式和(RSRS)-形式.(RSSR)-形式是相邻两个磷原子的甲基朝向大环膦形成的平面四方形之上,而另外两个相邻磷原子的甲基朝向平面四方形之下.(RSRS)-形式是4个磷原子的甲基都在平面四方形之上[21].立体异构体的存在会给产物分离提纯带来困难,也会增加后续反应研究的复杂度.
本研究选择第六副族过渡金属Mo作为模板,在其上构筑不含异构体的平面大环四齿膦配体.不论是在配体前体的合成还是在模板法关闭大环的过程中,该方法都能克服膦分子的手性问题,简化分离提纯的步骤.
1 实验部分
1.1 仪器与药品
实验中所有试剂均为分析纯.本实验过程除了季钅粦盐的提纯过程,大部分是在严格无水无氧条件下操作的,1,5-二磷杂双环[3.3.0]辛烷(化合物A)[22]按照文献报道的方法制备.其他试剂通过商业途径购买,如没有特别说明,所使用的试剂已经过除气和除水处理,固体药品均经过除气操作.
核磁共振谱均在Bruker 400 MHz核磁共振仪上于合适的溶剂中测试,化学位移参考残留溶剂(1H)或外标(31P{1H}:85% H3PO4).单晶数据通过在Bruker Smart Apex-Ⅱ上进行X射线单晶衍射分析得到.QEplus-ESI-MS的测量在Q Exactive高分辨率液相色谱-质谱仪Q Exactive Plus上进行.元素分析测试在UNICUBE元素分析仪上进行.
1.2 开链四齿膦配体L1的合成
化合物A的DMF溶液(3.9 mmol,0.02 mol/L)与1,3-二碘丙烷(1.95 mmol,226 μL)在DMF(15 mL)中加热到50 ℃反应24 h,析出大量白色固体,即化合物B[23-25],乙醚洗涤白色固体,减压抽干溶剂得到纯净化合物B,产率87%(1.7 mmol).将乙醚蒸气扩散至化合物B的DMF饱和溶液中,生长出无色针状晶体.1H NMR(400 MHz,C2D6SO):δ=2.70(dt,2JH—H=15.1 Hz,3JH—H=7.4 Hz,4H),2.57~2.47(m,4H),2.39~2.28(m,4H),2.12~1.81(m,18H).31P{1H} NMR(162 MHz,C2D6SO):δ=83.4(d,1JP—P=242.6 Hz,2P),-55.2(d,1JP—P=242.7 Hz,2P).高分辨质谱(QEplus-ESI,m/z):[C15H30P4]2+理论值167.064 4,实测值167.063 9.元素分析C15H30I2P4:理论值C 30.63,H 5.14,实测值C 31.76,H 5.34.
在100 mL的史莱克管中混合化合物B(435 mg,0.74 mmol)和THF(10 mL),加入二异丁基氢化铝(DIBAL)的正己烷溶液(2.96 mL,1 mol/L,2.96 mmol)室温搅拌30 min后,用甲醇淬灭至无气泡冒出,减压抽干溶剂,正己烷提取得到顺式开链四齿膦配体L1,产率10%(0.07 mmol).31P{1H} NMR(162 MHz,正己烷):δ=-30.2(s,2P),-67.7(s,2P).31P NMR(162 MHz,正己烷):δ=-30.2(伪s,2P),-67.5(伪d,1JP—H=199.8 Hz,2P).
1.3 关环前体的合成
将Mo(O)Cl2(PMePh2)3(0.07 mmol,54.9 mg)(M)[26-27]和配体L1的THF溶液(0.07 mmol,0.002 mol/L)在THF(10 mL)中混合,绿色固体逐渐溶解,且析出紫色固体.将紫色固体用THF洗涤后,再用乙腈提取,得到M(Cl)L1的乙腈溶液,将乙醚蒸气扩散至M(Cl)L1的饱和乙腈溶液中得到紫色块状晶体,产率71%(0.05 mmol).1H NMR(400 MHz,CD3CN):δ=6.19(d,1JP—H=344.2 Hz,2H,P—H),2.54~1.87(m,30H).31P{1H} NMR(162 MHz,MeCN):δ=3.3~1.8(m,AA′XX′,2P),-34.3~-35.7(m,AA′XX′,2P).31P NMR(162 MHz,MeCN):δ=2.6(伪d,2JP—P=111.3 Hz,2P),-35.0(伪dd,1JP—H=343.1 Hz,2JP—P=120.7 Hz,2P,P—H).高分辨质谱(QEplus-ESI,m/z):[C15H32ClMoOP4]+理论值485.014 6,实测值485.012 0.元素分析C15H32Cl2MoOP4:理论值C 34.70,H 6.21,实测值C 30.45,H 6.14.
在室温下,将M(Cl)L1(20.8 mg,0.04 mmol)溶于甲醇,加入1倍量的MeOLi(1.5 mg,0.04 mmol),紫色溶液立即变黄,减压抽出挥发物,用二氯甲烷提取残余物,减压抽干得到黄色固体粉末M(OMe)L1,分离产率90%(0.036 mmol).1H NMR(400 MHz,CD3CN):δ=5.77(d,1JP—H=315.5 Hz,2H,P—H),3.10(br,s,3H,OCH3),2.51~1.55(m,30H).31P{1H} NMR(162 MHz,DCM):δ=3.8~1.9(m,AA′XX′,2P),-33.6~-35.4(m,AA′XX′,2P).31P NMR(162 MHz,DCM):δ=2.9(伪d,2JP—P=180.0 Hz,2P),-34.5(伪dd,1JP—H=302.8 Hz,2JP—P=190.2 Hz,2P,P—H).高分辨质谱(QEplus-ESI,m/z):[C16H35MoO2P4]+理论值481.063 6,实测值481.062 5.元素分析C16H35ClMoO2P4:理论值C 37.33,H 6.85,实测值C 37.53,H 6.88.
1.4 烷基化合成平面大环四齿膦配体
将M(OMe)L1(0.02 mmol,10.3 mg)和THF(7 mL)混合,降温至-50 ℃,加入2倍量KHMDS(0.04 mmol,40 μL),黄色固体溶解,液相变成墨绿色;继续在-50 ℃低温条件下加入邻二溴苄(0.02 mmol,5.3 mg),立刻产生黄色沉淀,液相也变成黄色;在缓慢恢复至室温的过程中,析出大量橙黄色固体,室温反应12 h,橙黄色沉淀量增多,液相颜色变浅.先用THF洗涤橙黄色固体,再用乙腈提取关环产物,用KOTf(3.8 mg,0.02 mmol)将关环产物外界的阴离子交换成三氟甲磺酸根阴离子,过滤后收集滤液,真空除去溶剂,残余物用THF洗涤,然后干燥得到M(OMe)L2.分离产率50%(0.01 mmol).1H NMR(400 MHz,CD3OD):δ=7.24~7.20(br,m,4H,CHPh),3.26(br,s,3H,OCH3),2.19~2.04(br,m,34H).31P{1H} NMR(162 MHz,CD3CN):δ=3.2(br,s,4P).高分辨质谱(QEplus-ESI,m/z):[C24H41MoO2P4]+理论值583.110 6,实测值583.110 1.元素分析C25H41F3MoO5P4S:理论值C 41.10,H 5.66,实测值C 41.51,H 6.04.
2 结果与讨论
大环膦配体出现立体异构体的原因是磷原子四面体翻转困难,而侧环的引入可以使磷原子的取代基变成相同的,从而有效避免立体异构体的产生(图1).同时,侧环的引入也能增强配体整体的环刚性,让整个环状配体配位更加牢固.考虑到环尺寸的问题,两个侧环都选择八元环.配体的合成采用先合成开链四齿配体,然后与模板金属配位,最后通过关环反应构筑大环的过程.四齿配体配位点多,螯合效应强,对配位后的后续反应相对有利.使用Mo(O)Cl2(PMePh2)3作为金属模板,是利用了配位氧原子的反位效应,有利于开链四齿膦配体配位时处于赤道面上,从而得到最终期望的配位模式.
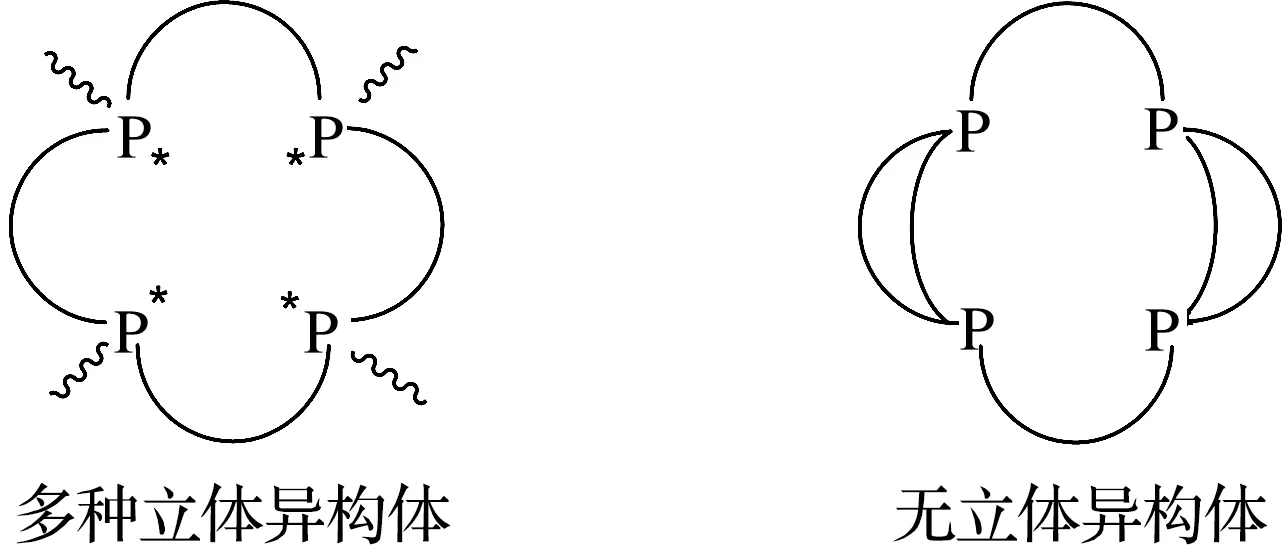
图1 大环四齿膦配体的立体化学结构
使用1,3-二碘丙烷将化合物A烷基化,以良好的收率合成了季钅粦盐二碘化物B(图2).季钅粦盐二碘化物B的磷谱在84.5和-54.0处出现两组互相耦合的多重峰(1JP—P=245.4 Hz),分别对应两种不同类型的磷原子.将乙醚蒸气扩散到化合物的DMF溶液中生长出适用于X射线单晶衍射分析的晶体(图3).用DIBAL还原化合物B会断开其中的P—P键,从而得到含有二级膦的开链四齿配体L1.L1的质子去耦磷谱在-30.2和-67.7处出现两个单峰.相应地,在质子耦合磷谱中,-67.7处的单峰变为双峰(1JP—H=199.8 Hz),这有力地证明了L1中含有P—H键.配体L1可以用正己烷从反应的残留物中提取出来,虽然有少量杂质,但对后续的配位反应并无显著影响.
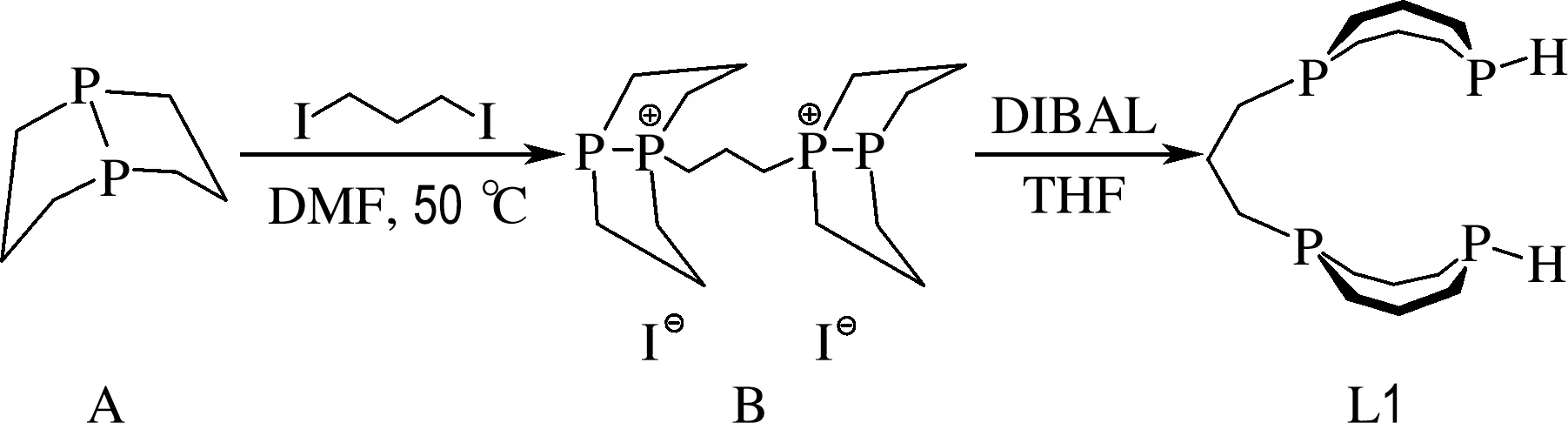
图2 开链四齿膦配体L1的合成
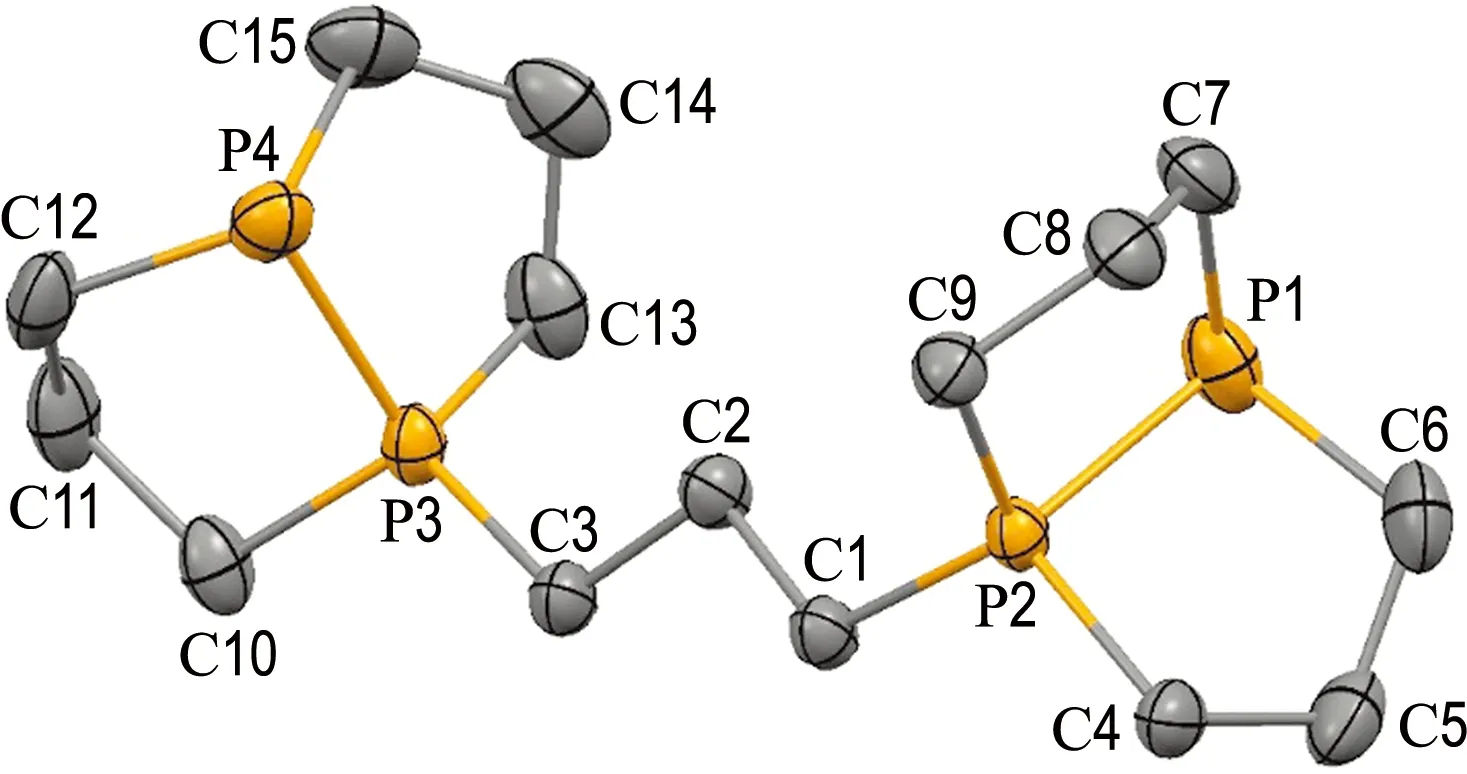
图3 化合物B(阳离子部分)的X射线单晶结构(CCDC:2171491)
在室温下将配体L1加入Mo(O)Cl2(PMePh2)3(M)的THF溶液中(图4),绿色溶液逐渐变为无色,且析出紫色固体.反应上清液的磷谱显示只有从中心金属上被取代下来的PMePh2,将紫色沉淀溶于乙腈,在质子去耦磷谱中出现两组互相耦合的多重峰,形成典型的AA′XX′自旋体系.同样,-35.0处的信号在质子耦合磷谱中P—H的耦合常数为1JP—H=343.1 Hz,表明该配合物中存在P—H键;因此推测合成了具有四齿膦配体的钼氧配合物(M(Cl)L1).将乙醚蒸气扩散到紫色沉淀的饱和乙腈溶液中,生长出M(Cl)L1的晶体.X射线晶体学研究揭示了Mo中心的伪八面体几何形状,其中四齿膦配体位于赤道平面上,氯离子处于氧原子的反位(图5).
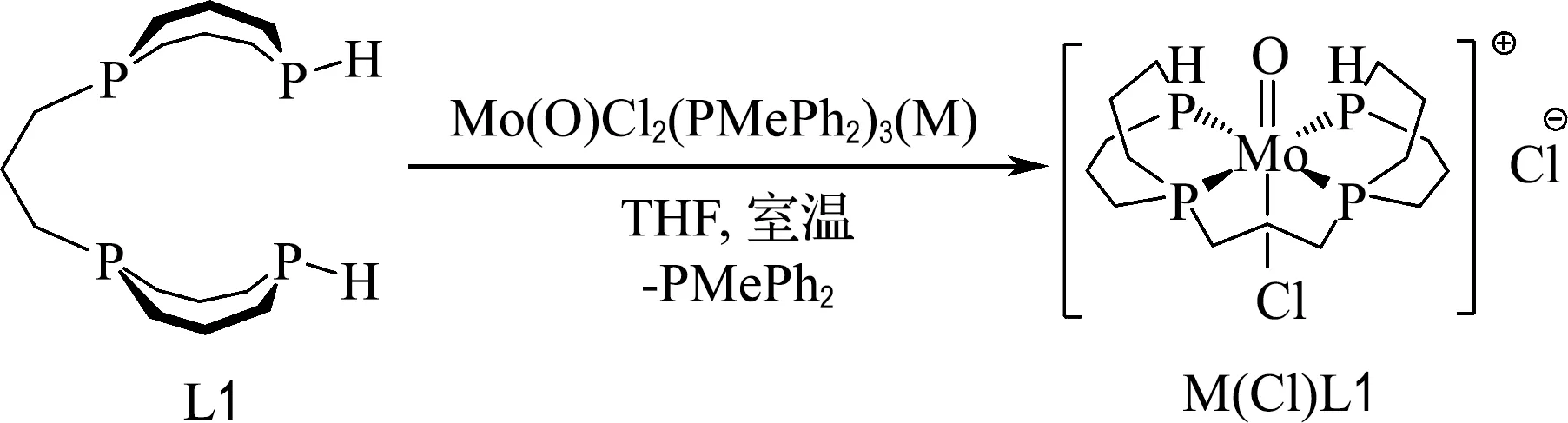
图4 M(Cl)L1的合成
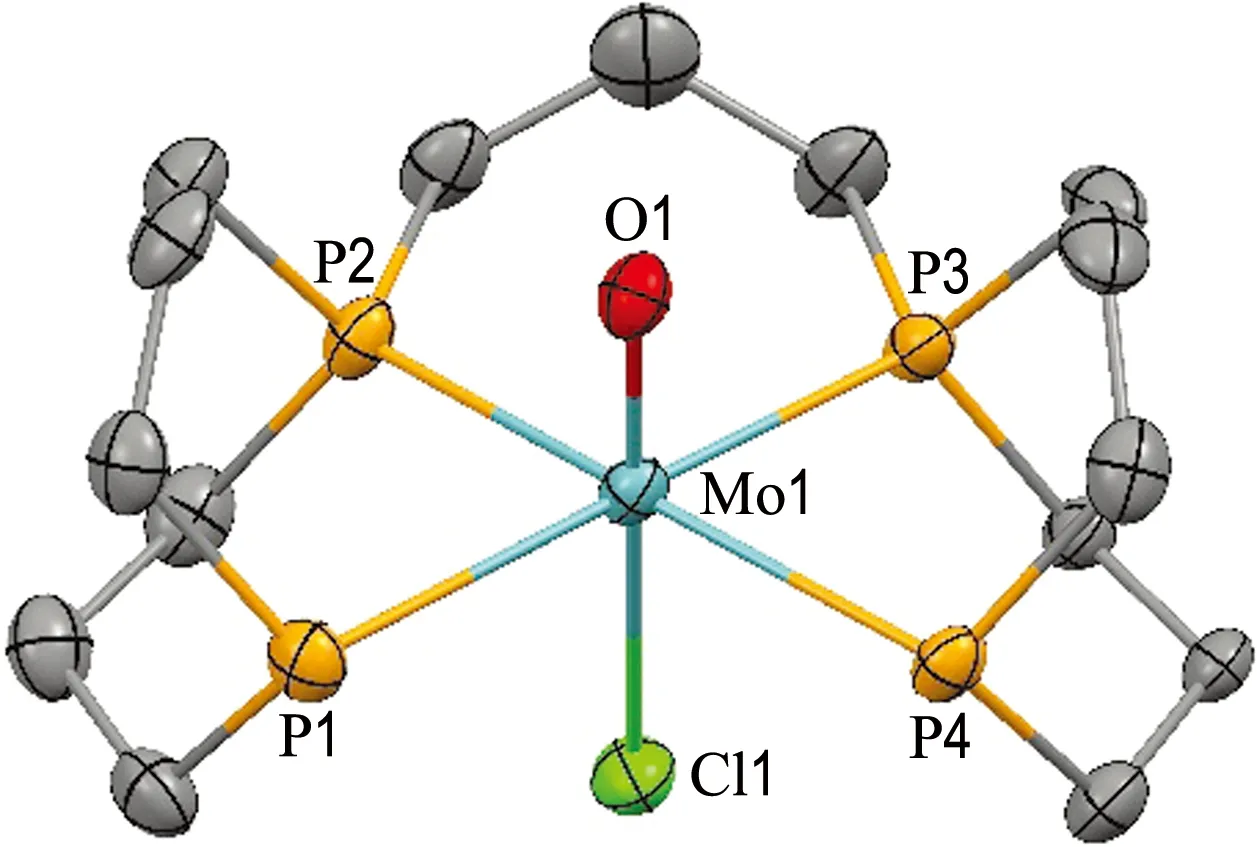
图5 配合物M(Cl)L1(阳离子部分)的X射线单晶结构(CCDC:2171492)
与Ni(Ⅱ)或Pd(Ⅱ)配合物中的二级膦配体可以用弱碱[12,28-29](如三乙胺和K2CO3)去质子化不同,配合物M(Cl)L1中的配体只能用较强的碱如KHMDS去质子化,尽管含Mo的部分在去质子化之前整体是阳离子,然而,M(Cl)L1似乎不利于碱性条件下的闭环反应,这可能是因为一旦去质子化,Mo—Cl键就会断裂,因为处于氧原子反位的Cl-是较好的离去基团,这导致配合物的结构发生变化.因此,将MeOLi加入M(Cl)L1的甲醇溶液中,把M(Cl)L1中的Cl-替换为MeO-,得到配合物M(OMe)L1.正如预期的那样,在-50 ℃下,在THF中用KHMDS将配合物M(OMe)L1去质子化,然后加入等量的邻二溴苄,析出的黄色固体即为大环膦的钼配合物(M(OMe)L2),收率良好(图6).配合物M(OMe)L2的磷谱在2.7处出现峰形很宽的峰包,并且在氢谱上,M(OMe)L2苯环上的氢原子和配体骨架上的氢原子都出现很宽的峰包.这一现象不太常见,可能是大环的构象限制导致的某些动力学效应引起,值得后续进一步研究,而这也证实了分子内大环结构的形成.配合物M(OMe)L2在高分辨质谱中于m/z=583.110 13处显示非常强的信号,与配合物阳离子的信号完全对应(图7).该处信号的同位素特征峰形也显示,此物质是一种单核钼配合物,与目标物质完全一致.
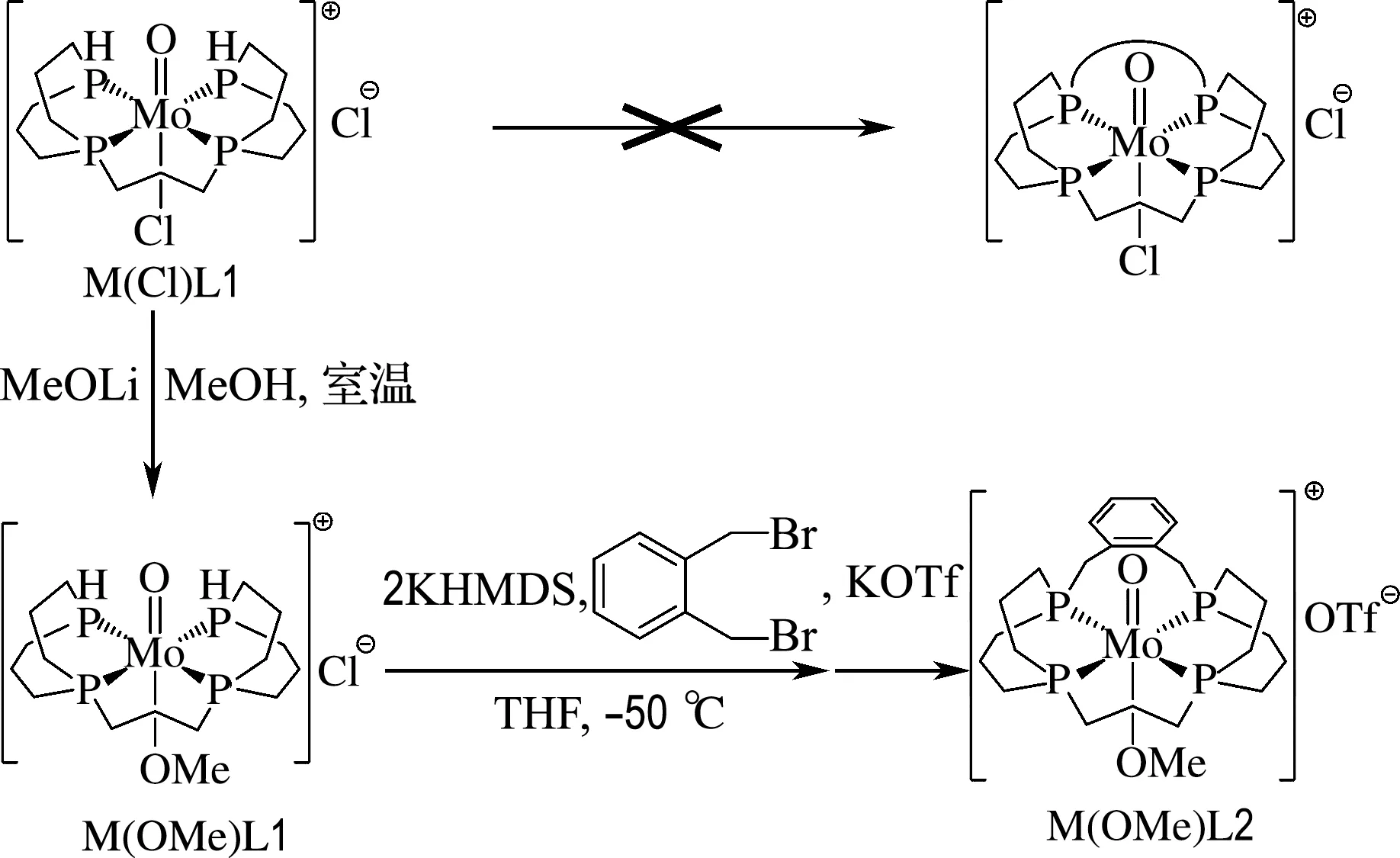
图6 平面大环四齿膦配体合成
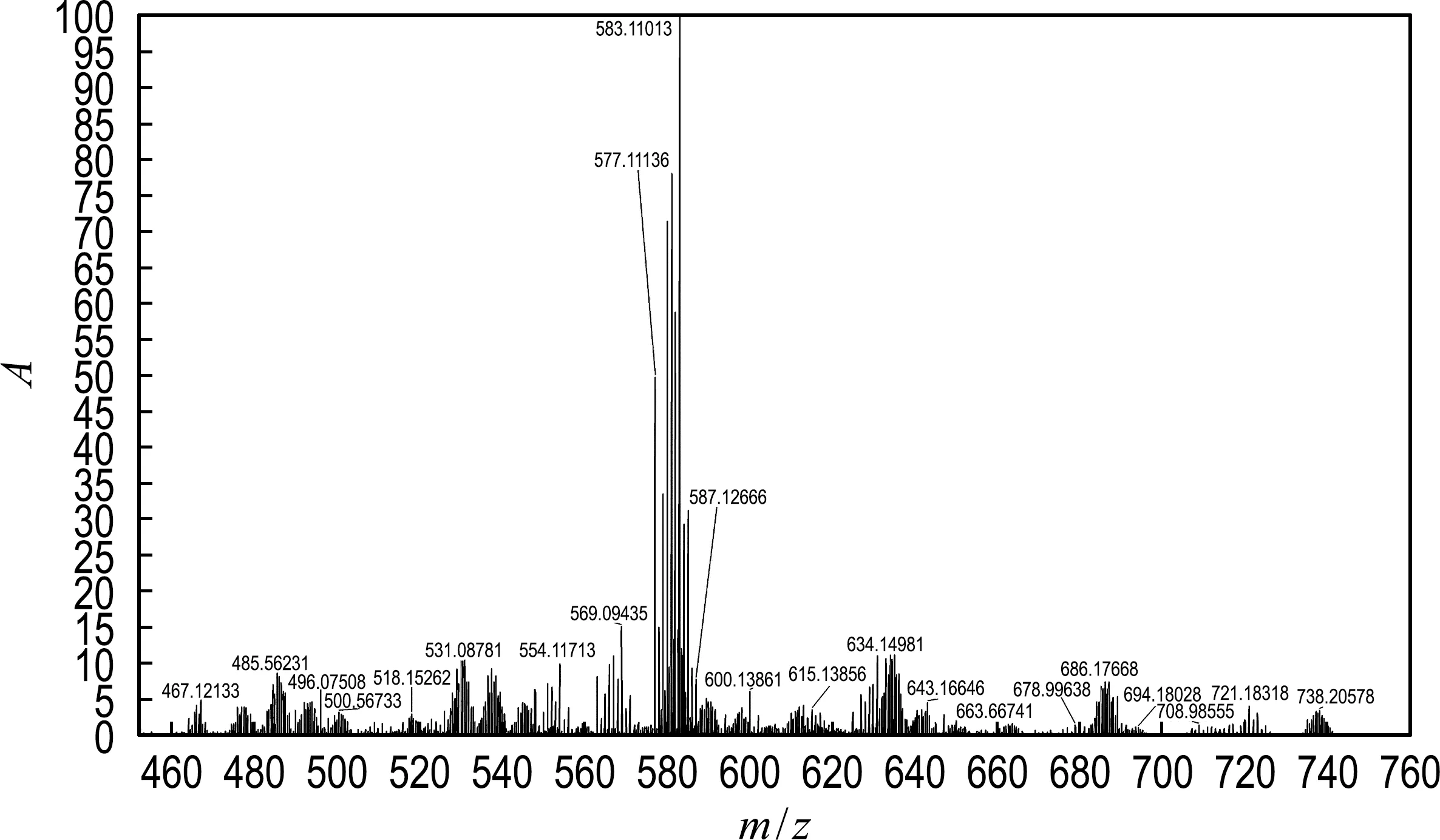
图7 M(OMe)L2在甲醇中的QEplus-ESI-MS谱图
3 结 语
首次以钼为中心金属,通过模板反应在其上合成了平面大环四齿膦配体,得到一种新型钼氧配合物.此配合物的大环膦配体结构中有两个侧环,在合成过程中避免了立体异构体的产生,能使分离提纯过程得到简化.目前正在进一步研究这种类型配体的钼氧配合物的反应性质.
