铁死亡中的溶酶体降解系统:质量调控与能量代谢*
2023-10-11刘师佐杨欢汤蓉王延蛟
刘师佐, 杨欢, 汤蓉, 王延蛟
铁死亡中的溶酶体降解系统:质量调控与能量代谢*
刘师佐1, 杨欢2, 汤蓉1, 王延蛟3△
(1新疆医科大学基础医学院,新疆 乌鲁木齐 830017;2新疆医科大学第二临床医学院,新疆 乌鲁木齐 830017;3新疆医科大学基础医学院生物化学与分子生物学教研室,新疆地方病分子生物学重点实验室,新疆 乌鲁木齐 830017)
铁死亡;自噬;溶酶体
铁死亡(ferroptosis)是近年来新发现的一种细胞死亡方式,其特征是胞内游离铁和脂质过氧化物的积累[1]。巨自噬(macroautophagy;下文简称自噬,autophagy)是隔离膜将胞质内底物包裹,并在溶酶体内降解为小分子以维持细胞发育和环境稳态的过程。自噬的质量调控和能量代谢对铁死亡的发生有重要意义。铁死亡中的质量调控由泛素系统和自噬-溶酶体系统构成,主要参与铁死亡相关蛋白的降解。一方面,自噬-溶酶体系统参与的质量调控影响铁的储存、输入和输出,也参与了游离脂质的产生和抗氧化物质的降解,有研究认为铁死亡是一种自噬依赖性细胞死亡(autophagy-dependent cell death, ADCD)[2]。另一方面,自噬或巨胞饮(macropinocytosis)等介导的能量代谢是氧化还原相关蛋白合成、线粒体代谢和脂质生成的基础,与铁死亡的发生密切相关[3]。能量代谢和质量控制均是细胞的重要功能,它们对铁死亡有何影响?二者之间存在何种关系?以溶酶体降解系统为基础的质量调控和能量代谢通常是相互统一的[4],但在铁死亡中却相反[2, 5-6]。在质量调控中,自噬、伴侣介导的自噬或非自噬途径通过降解储铁蛋白、脂滴、抗氧化物质或特定的细胞器来增加芬顿反应的底物,干扰能量代谢,从而促进铁死亡[6]。在能量代谢中,自噬或巨胞饮等通过增加铁死亡期间的氨基酸代谢、抗氧化蛋白的合成等来缓解细胞死亡[7]。因此,溶酶体及其调控信号在特定的条件下发挥双重作用,其既能通过对功能蛋白的选择性降解促进铁死亡,又能通过能量代谢抑制铁死亡。本文中,我们基于铁死亡中的溶酶体系统(包括自噬、伴侣介导的自噬、自噬非依赖性溶酶体降解和巨胞饮,其中主要以自噬为主),阐述了铁死亡中质量调控与能量代谢之间的关系,并基于目前铁死亡的临床应用提出待解决的问题。
1 溶酶体的质量调控促进铁死亡
溶酶体在铁死亡中参与质量调控的主要途径有:(1)选择性自噬,在生理或病理条件下,自噬受体与底物特异性结合并被自噬体吞噬,最终被溶酶体降解;(2)伴侣介导的自噬,伴侣热休克同源蛋白70(heat shock cognate protein 70, HSC70)与底物结合并将其递送到溶酶体最终被分解[8];(3)自噬非依赖性溶酶体降解,指不依赖于自噬或伴侣介导的自噬机制,由其他分子介导底物进入溶酶体被降解的过程[9]。质量调控自噬是促进铁死亡进行的主要因素[10],与以下三点有关。
1.1参与铁的调控铁是铁死亡发生的基础。在衰老细胞中,自噬受损导致细胞内铁含量高达正常水平的30倍[11]。自噬参与铁的储存、输入和输出[12-13]。核受体辅激活蛋白4(nuclear receptor coactivator 4, NCOA4)是调控铁蛋白自噬的主要分子,它与储铁蛋白结合并将其募集于自噬溶酶体中降解(铁蛋白自噬),导致Fe2+的释放,促进铁死亡[14]。NCOA4含有多个泛素化位点,TRIM7 (tripartite motif containing 7)通过K48直接结合NCOA4并将其泛素化降解,抑制铁蛋白降解,敲除能够抑制铁死亡[15]。在缺氧条件下,miR-6862-5p与NCOA4 mRNA结合抑制其翻译,维持线粒体铁蛋白稳定并促进缺氧应激条件下的线粒体代谢[16]。在铁死亡过程中,线粒体可以作为游离Fe2+的缓冲区以降低Fe2+对细胞的毒性,因此自噬仅对铁蛋白的降解不会显著促进铁死亡。Thiamet G通过促进-GlcNAc糖基化不仅进一步加强铁蛋白自噬,还促进线粒体裂解和线粒体自噬以增加游离Fe2+的来源(图1A)[17]。目前自噬与铁的输入之间的关系存在争议。有研究表明,自噬的激活还能够上调转铁蛋白受体1(transferrin receptor 1, TFR1)表达,增加细胞对铁的吸收[18]。但也有研究表明,自噬受体p62与TFR1结合介导其降解,降低细胞对铁死亡的敏感性[19]。此外,p62与铁输出蛋白SLC40A1/ferroportin-1结合,介导其被选择性降解[20]。
1.2促进不饱和脂肪酸积累脂滴是细胞内储存脂质的细胞器,脂滴分解产生的脂质参与多种代谢途径,不饱和脂肪酸积累是铁死亡发生的前提,其与活性氧(reactive oxygen species, ROS)反应生成脂质过氧化物[21]。黄体酮受体膜成分1(progesterone receptor membrane component 1, PGRMC1)是一种血红素结合蛋白,其过表达能改变脂代谢,促进肿瘤细胞增殖。在紫杉醇耐药的头颈癌中,PGRMC1的过表达通过Sirt1-AMP活化蛋白激酶(AMP-activatited protein kinase, AMPK)轴诱导RAB7A依赖性脂肪自噬[22],使用erastin能够有效抑制肿瘤的生长[23](图1B)。神经胶质成熟因子β(glia maturation factor-β, GMFB-β)在糖尿病视网膜病变中上调,介导ATP酶ATP6V1A易位至溶酶体表面并使其碱化,功能障碍的溶酶体使ACSL4 (acyl-CoA synthetase long-chain family member 4)积累,后者促进不饱和脂肪酸的产生,诱导铁死亡的进行[9]。
1.3抑制谷胱甘肽过氧化物酶4(glutathione peroxidase 4, GPX4)GPX4利用还原型谷胱甘肽(reduced glutathione, GSH)将毒性脂质过氧化物转化为无毒脂质醇,是细胞抵抗铁死亡的关键酶[24]。Wu等[25]发现,erastin通过上调溶酶体相关膜蛋白2a和伴侣HSC70促进伴侣介导的自噬以降解GPX4,促进过氧化物的产生导致铁死亡。Xue等[26]发现外源Cu2+与GPX4的Cys107和Cys148直接结合以促进GPX4泛素化和GPX4聚集体的形成,自噬受体Tax1结合蛋白1随后促进GPX4降解和铁死亡。Liu等[27]发现,作为自噬诱导信号的哺乳动物雷帕霉素靶蛋白(mammalian target of rapamycin, mTOR)被雷帕霉素抑制后能介导自噬依赖性GPX4降解,而过表达GPX4能促进mTOR的活化以抑制自噬,缓解铁死亡的发生。同样作为自噬诱导信号,AMPK介导beclin-1与溶质载体家族7成员11(solute carrier family 7 member 11, SLC7A11)结合抑制系统Xc-,后者为GPX4的合成提供关键的半胱氨酸,导致抗氧化物质合成不足,最终促进铁死亡[28]。然而,也有研究表明在Fin56通过mTOR非依赖性途径介导铁死亡中GPX4的自噬降解,但mTOR抑制剂却增强膀胱癌细胞对Fin56的敏感性[29](图1C)。
长期以来,ADCD被认为是由于过度自噬导致细胞内容物无差别的降解,其无法解释铁死亡中的质量调控自噬。铁死亡中的自噬为我们阐明了ADCD也存在精细的调控机制,这种灵敏的选择性降解功能参与细胞器的调节和各种蛋白的表达,并通过促进脂质的产生、抑制抗氧化物质的形成而导致铁死亡。然而,关于ADCD在铁死亡中的调节机制尚不明确。是什么决定了自噬的性质?哪些分子特异性参与了该过程?细胞器的损伤或功能紊乱(如脂滴[22]、线粒体[30]、内质网[31]或过氧化物酶体[32])可能是导致铁死亡的原因之一,选择性自噬对细胞器的降解起到何种作用?
2 溶酶体的能量代谢缓解铁死亡
半胱氨酸饥饿诱导铁死亡的细胞中ATP利用率显著降低,表明铁死亡与能量代谢密切相关[33]。BAY-876和多柔比星组成的纳米颗粒通过抑制葡萄糖转运蛋白1的功能以限制葡萄糖摄取,同时多柔比星降低ATP在胞内水平,诱导肿瘤细胞铁死亡[34]。线粒体中的谷氨酰胺代谢产生的α-酮戊二酸参与三羧酸循环,与电子传递链共同促进铁死亡[35]。人铁质反射素2(sideroflexin 2, SFXN2)是一种参与线粒体铁代谢的线粒体外膜蛋白,在多发性骨髓瘤中,SFXN2与血红素加氧酶1结合抑制饥饿诱导的自噬/线粒体自噬来减轻铁死亡[36]。
Li等[37]发现,在多种铁死亡诱导模型中,葡萄糖饥饿通过激活AMPK抑制铁死亡,表明这种机制可能具有普遍性。AMPK被肝激酶B1(liver kinase B1,LKB1)磷酸化,以抑制乙酰辅酶A羧化酶(acetyl-CoA carboxylase, ACC)的磷酸化。ACC是脂肪酸生物合成的限速酶,AMPK的磷酸化降低脂肪酸合成和脂质过氧化[38]。在敲除单羧酸转运蛋白4(monocarboxylate transporter 4,)的膀胱癌细胞中,p-AMPK显著下调,ACC表达量增高,并且能量代谢自噬被抑制导致铁死亡的发生(图1D)[39]。同时,在特定生物学条件下,p-AMPKα2激活能量代谢自噬以维持氧化还原平衡、线粒体功能和脂代谢等,缓解小鼠心肌细胞的铁死亡[5]。
mTOR是调控自噬的另一个营养感受器,也是溶酶体发挥功能的重要调节器。在铁死亡中,mTOR的不同状态能够决定细胞的存活。mTOR被激活后促进蛋白质的合成代谢,是细胞中最耗能的过程之一;当细胞内氨基酸、核苷酸等小分子不足,mTOR被抑制促进分解代谢,如自噬或巨胞饮作用[40]。在胱氨酸饥饿条件下,mTOR被抑制,综合应激反应(integrated stress response)被激活,介导下游的各种生物过程促进胱氨酸的合成代谢[41]。在多数情况下,细胞主要通过营养转运蛋白从细胞外摄取半胱氨酸,如胱氨酸/谷氨酸转运蛋白SLC7A11,摄取的胱氨酸被转化为半胱氨酸,后者是GSH的重要底物。半胱氨酸的缺乏导致GSH的合成减少,细胞无法有效抑制脂质ROS的产生,最终导致铁死亡。Conlon等[42]通过高通量药物筛选发现mTOR抑制剂能有效缓解铁死亡,表明氨基酸等小分子的合成在铁死亡期间尤为重要。Byun等[43]随后发现,在索拉非尼耐药的肝癌细胞中,线粒体功能障碍会激活PI3K-RAC1-PAK1来促进巨胞饮作用,增加癌细胞在胱氨酸剥夺条件下对外周白蛋白的摄取。这种含丰富半胱氨酸的蛋白被癌细胞摄取后,mTOR信号被抑制以增强溶酶体对白蛋白的分解能力,经组织蛋白酶B(cathepsin B, CTSB)分解后由胱氨酸转运体(cystinosin, CTNS)输出至胞质,促进GSH的合成(图1E)[44]。mTOR的激活在特定条件下也能抵抗铁死亡。GPX4是一种硒蛋白,合成效率低且耗能极大。当胱氨酸充足时,细胞激活Rag-mTORC1-4EBP轴促进GPX4合成,抑制脂质过氧化,这也符合当ATP利用率低促进铁死亡的理论[45]。总之,在底物匮乏时,mTOR受抑制以介导半胱氨酸的生成,而在底物充足、能量丰富的情况下,mTOR被激活以促进GPX4的转录翻译,mTOR不同状态可能是细胞整合当时环境对物质的需求而做出不同选择的结果。因此,确定当下环境中细胞对底物(半胱氨酸)或产物(GPX4/GSH)的需求,靶向mTOR来调控铁死亡是临床治疗的新思路。
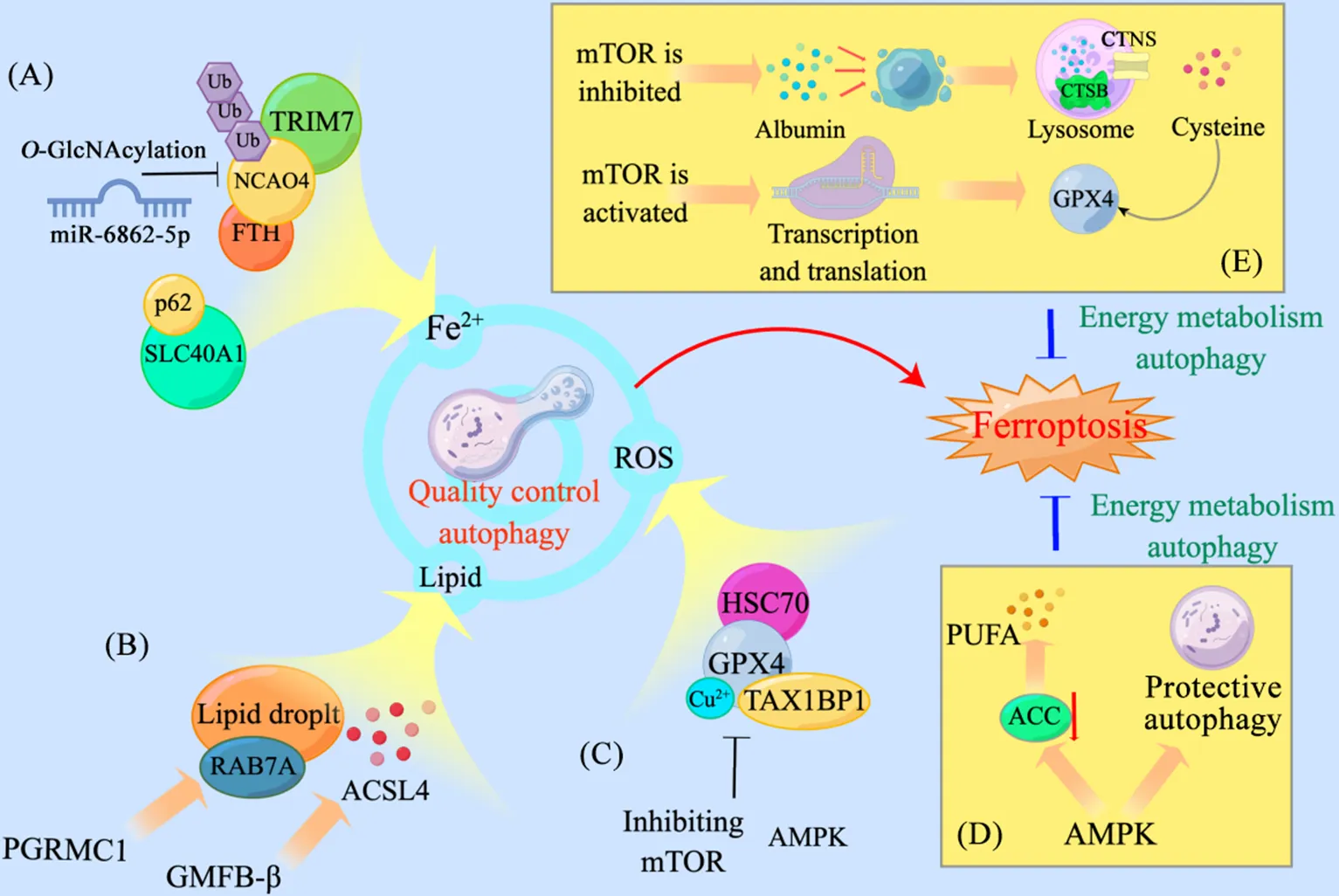
Figure 1. Quality control and energetic metabolism of autophagy regulatory pathways for ferroptosis. A: nuclear receptor coactivator 4 (NCOA4) is regulated by ubiquitination and O-GlcNAcylation to influence free Fe2+ production, and p62 binding to solute carrier family 40 member 1 (SLC40A1/ferroportin-1) promotes Fe2+ accumulation; B: glia maturation factor-β (GMFB-β) promotes RAB7A-dependent lipophagy for lipogenesis, and progesterone receptor membrane component 1 (PGRMC1) promotes accumulation of acyl-CoA synthetase long-chain family member 4 (ACSL4); C: heat shock cognate protein 70 (HSC70), Cu2+ or Tax1-binding protein 1 (TAX1BP1) binding to glutathione peroxidase 4 (GPX4) promotes GPX4 degradation, and activation of AMP-activatited protein kinase (AMPK) or inhibition of mammalian target of rapamycin (mTOR) also promotes the degradation of GPX4; D: AMPK activates energy metabolism autophagy while suppressing acetyl-CoA carboxylase (ACC) expression; E: in the absence of substrate for GPX4 synthesis, mTOR is inhibited to increase the uptake of extracellular albumin, which is catabolized by lysosomes to produce cysteine, the raw material for GPX4 synthesis; when substrate is sufficient, mTOR is activated to promote transcriptional translation of GPX4. TRIM7: tripartite motif containing 7; Ub: ubiquitin; PUFA: polyunsaturated fatty acid; CTSB: cathepsin B; CTNS: cystinosin; ROS: reactive oxygen species. Some cartoon components were from www.figdraw.com for model drawing.
3 铁死亡中的质量调控与能量代谢
根据细胞的生理需求,自噬主要分为质量调控自噬和能量代谢自噬。前者需多种分子机器的相互作用,为选择性自噬;而后者无差别地回收降解底物,通常认为其为非选择性自噬。绝大多数情况下,两种类型的自噬是统一的过程,如在激活能量代谢自噬的同时,自噬受体会与损伤的细胞器结合(如去极化的线粒体),促进细胞代谢的同时维持质量调控[4]。然而在铁死亡中,二者功能并不统一。在敲除的肺癌中,自噬促进线粒体呼吸和ATP的产生,而抑制自噬并使用曲美替尼处理导致细胞铁死亡[46];抑制保护性自噬可增加胶质母细胞瘤对替莫唑胺的敏感性[47],而多数研究认为选择性自噬促进铁死亡[2, 14, 17, 26],但其促进机制尚不清楚。AMPK和mTOR是自噬的主要激活信号,部分研究却显示自噬能够独立于mTOR信号介导铁死亡[47],但使用mTOR抑制剂能够阻断铁死亡的进行[42]。ATG9A是一种多跨膜蛋白,在营养匮乏时参与自噬体膜的成熟。Liu等[48]指出,铁死亡诱导TMEM164 (transmembrane protein 164)表达,通过调控ATG5-ATG12-ATG16L1复合物促进自噬体的形成,而不是ATG9A。铁死亡与饥饿条件下诱导的自噬可能并非同一套机制,因此在不同的生物学条件下,自噬在铁死亡中同样存在两种形式:以分解代谢为主,为细胞提供能量或氨基酸的保护性自噬;以质量调控为主,自噬选择性降解铁死亡抑制分子的致死性自噬。
此前有研究表明,保护性自噬和致死性自噬同时存在于细胞中[49],二者的平衡状态决定细胞的生存结果。因此,溶酶体作为细胞的“回收站”,可能参与保护性和致死性自噬过程中相关生物分子的降解。在能量应激期间,巨胞饮和自噬共同抑制铁死亡,二者的主要区别在于前者通过摄取细胞外白蛋白至溶酶体,分解产生半胱氨酸以提供GSH的原料,后者主要控制细胞内能量代谢、脂代谢等抑制铁死亡;而二者有一个共同的降解系统——溶酶体。Armenta等[44]的研究提示,通过靶向与自噬/巨胞饮相关的溶酶体中60余种酸性水解酶,能平衡保护性与致死性自噬来调节铁死亡,对相关疾病的治疗可能具有重要意义。
4 总结与展望
本文基于细胞中的溶酶体降解系统,以自噬为主阐述其在铁死亡中的作用。自噬在铁死亡中有两种作用方式——质量调控和能量代谢,前者通过降解特定蛋白(铁代谢相关蛋白)、细胞器(脂滴、线粒体或内质网等)促进铁死亡,后者通过调控氨基酸代谢、脂质代谢等抑制铁死亡。因此,鉴别特定条件下自噬的性质,对临床肿瘤治疗有关键意义。
目前,自噬靶向治疗在临床中的应用存在两道屏障:(1)肿瘤中自噬的性质不能得到准确判断,其中包括鉴别单一性自噬和双重性自噬。单一性自噬,即自噬在组织中只具备促进细胞生存或死亡的能力,而双重性自噬指的是自噬既能促进细胞生存,也能促进细胞死亡,最后细胞的生存状态取决于二者的平衡。对于单一性自噬,目前已经发现部分标志性分子(如β-thujaplicin[50]、GBA1[51]等)能够诱导ADCD,但其敏感性和特异性仍有待考量,应用于临床进行分子诊断仍不成熟。对于双重性自噬,部分临床药物可诱导多种死亡模式促进癌细胞死亡。如在索拉菲尼处理的肝癌细胞中,自噬被激活以抑制细胞凋亡,介导细胞化疗耐药[52],但其又能诱导肝癌细胞自噬依赖性铁死亡[31],那么自噬如何调控细胞的生存与死亡?抑制自噬对细胞敏感性有何影响?探索双重性自噬介导细胞生与死的“临界点”,或寻找并量化不同性质自噬的标志分子,以确定对细胞的存活状态占主导作用的自噬,可能是解决该问题的策略之一。(2)适用于临床的自噬靶向药物仍未出现。由FDA批准的自噬抑制剂氯喹和羟氯喹需要高浓度才能够抑制自噬,目前绝大多数仅适用于肿瘤的辅助治疗[53]。适用于临床自噬靶向药物的研发有3个不可避免的问题需要解决:其一,确定特异性作用于自噬机制的分子。由于大量的自噬分子均可独立于自噬发挥作用,因此选择在人体中稳定调控自噬的分子是目前最大的难题[54]。其二,解决化合物溶解度低、特异性差或效力弱等问题。其三,对于理想的化合物,应进一步探索其对凋亡、坏死、炎症等其他信号通路的作用,减少其对自噬调节的作用。因此,探索铁死亡中自噬的调控机制,寻找介导保护性自噬与致死性自噬的分子,明确二者的作用机制,寻找能够应用于临床的标志物,对人类疾病的预防与治疗有潜在意义。
[1]康传杰,张相彤,马威,细胞铁死亡发生与调控机制的研究进展[J]. 中国病理生理杂志, 2017, 33(3):567-571.
Kang CJ, Zhang XT, Ma W. Progress in occurrence and development of ferroptosis[J]. Chin J Pthophysiol, 2017, 33(3):567-571.
[2] Gao M, Monian P, Pan Q, et al. Ferroptosis is an autophagic cell death process[J]. Cell Res, 2016, 26(9):1021-1032.
[3] Stockwell BR. Ferroptosis turns 10: emerging mechanisms, physiological functions, and therapeutic applications[J]. Cell, 2022, 185(14):2401-2421.
[4] Deretic V, Kroemer G. Autophagy in metabolism and quality control: opposing, complementary or interlinked functions?[J]. Autophagy, 2022, 18(2):283-292.
[5] He H, Wang L, Qiao Y, et al. Epigallocatechin-3-gallate pretreatment alleviates doxorubicin-induced ferroptosis and cardiotoxicity by upregulating AMPKα2 and activating adaptive autophagy[J]. Redox Biol, 2021, 48:102185.
[6] Liu J, Kuang F, Kroemer G, et al. Autophagy-dependent ferroptosis: machinery and regulation[J]. Cell Chem Biol, 2020, 27(4):420-435.
[7] Yao X, Li W, Fang D, et al. Emerging roles of energy metabolism in ferroptosis regulation of tumor cells[J]. Adv Sci (Weinh), 2021, 8(22):e2100997.
[8] Cuervo AM, Wong E. Chaperone-mediated autophagy: roles in disease and aging[J]. Cell Res, 2014, 24(1):92-104.
[9] Liu C, Sun W, Zhu T, et al. Glia maturation factor-β induces ferroptosis by impairing chaperone-mediated autophagic degradation of ACSL4 in early diabetic retinopathy[J]. Redox Biol, 2022, 52:102292.
[10] Chen X, Yu C, Kang R, et al. Cellular degradation systems in ferroptosis[J]. Cell Death Differ, 2021, 28(4):1135-1148.
[11] Masaldan S, Clatworthy SAS, Gamell C, et al. Iron accumulation in senescent cells is coupled with impaired ferritinophagy and inhibition of ferroptosis[J]. Redox Biol, 2018, 14:100-115.
[12] Zhang S, Xin W, Anderson GJ, et al. Double-edge sword roles of iron in driving energy production versus instigating ferroptosis[J]. Cell Death Dis, 2022, 13(1):40.
[13] 闻照凤,李延莉,铁蛋白自噬在癌症发生发展中作用的研究进展[J]. 中国病理生理杂志, 2022, 38(7):1340-1344.
Wen ZF, Li YL. Role of ferritinophagy in occurrence and development of cancers[J]. Chin J Pathophysiol, 2022, 38(7):1340-1344.
[14] Mancias JD, Wang X, Gygi SP, et al. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy[J]. Nature, 2014, 509(7498):105-109.
[15] Li K, Chen B, Xu A, et al. TRIM7 modulates NCOA4-mediated ferritinophagy and ferroptosis in glioblastoma cells[J]. Redox Biol, 2022, 56:102451.
[16] Fuhrmann DC, Mondorf A, Beifuß J, et al. Hypoxia inhibits ferritinophagy, increases mitochondrial ferritin, and protects from ferroptosis[J]. Redox Biol, 2020, 36:101670.
[17] Yu F, Zhang Q, Liu H, et al. Dynamic-GlcNAcylation coordinates ferritinophagy and mitophagy to activate ferroptosis[J]. Cell Discov, 2022, 8(1):40.
[18] Park E, Chung SW. ROS-mediated autophagy increases intracellular iron levels and ferroptosis by ferritin and transferrin receptor regulation[J]. Cell Death Dis, 2019, 10(11):822.
[19] Shan J, Jiang W, Chang J, et al. NUF2 drives cholangiocarcinoma progression and migration via inhibiting autophagic degradation of TFR1[J]. Int J Biol Sci, 2023, 19(5):1336-1351.
[20] Li J, Liu J, Xu Y, et al. Tumor heterogeneity in autophagy-dependent ferroptosis[J]. Autophagy, 2021, 17(11):3361-3374.
[21] Liang D, Minikes AM, Jiang X. Ferroptosis at the intersection of lipid metabolism and cellular signaling[J]. Mol Cell, 2022, 82(12):2215-2227.
[22] Bai Y, Meng L, Han L, et al. Lipid storage and lipophagy regulates ferroptosis[J]. Biochem Biophys Res Commun, 2019, 508(4):997-1003.
[23] You JH, Lee J, Roh JL. PGRMC1-dependent lipophagy promotes ferroptosis in paclitaxel-tolerant persister cancer cells[J]. J Exp Clin Cancer Res, 2021, 40(1):350.
[24] Forcina GC, Dixon SJ. GPX4 at the crossroads of lipid homeostasis and ferroptosis[J]. Proteomics, 2019, 19(18):e1800311.
[25] Wu Z, Geng Y, Lu X, et al. Chaperone-mediated autophagy is involved in the execution of ferroptosis[J]. Proc Natl Acad Sci U S A, 2019, 116(8):2996-3005.
[26] Xue Q, Yan D, Chen X, et al. Copper-dependent autophagic degradation of GPX4 drives ferroptosis[J]. Autophagy, 2023, 19(7):1982-1996.
[27] Liu Y, WangY, Liu J, et al. Interplay between mTOR and GPX4 signaling modulates autophagy-dependent ferroptotic cancer cell death[J]. Cancer Gene Ther, 2021, 28(1/2):55-63.
[28] Song X, Zhu S, Chen P, et al. AMPK-mediated BECN1 phosphorylation promotes ferroptosis by directly blocking system Xc-activity[J]. Curr Biol, 2018, 28(15):2388-2399.e5.
[29] Sun Y, Berleth N, Wu W, et al. Fin56-induced ferroptosis is supported by autophagy-mediated GPX4 degradation and functions synergistically with mTOR inhibition to kill bladder cancer cells[J]. Cell Death Dis, 2021, 12(11):1028.
[30] Tadokoro T, Ikeda M, Ide T, et al. Mitochondria-dependent ferroptosis plays a pivotal role in doxorubicin cardiotoxicity[J]. JCI Insight, 2020, 5(9):e132747.
[31] Liu Z, Ma C, Wang Q, et al. Targeting FAM134B-mediated reticulophagy activates sorafenib-induced ferroptosis in hepatocellular carcinoma[J]. Biochem Biophys Res Commun, 2022, 589:247-253.
[32] Garcia-Bermudez J, Baudrier L, Bayraktar EC, et al. Squalene accumulation in cholesterol auxotrophic lymphomas prevents oxidative cell death[J]. Nature, 2019, 567(7746):118-122.
[33] Gao M, Monian P, Quadri N, et al. Glutaminolysis and transferrin regulate ferroptosis[J]. Mol Cell, 2015, 59(2):298-308.
[34] Jiang W, Luo X, Wei L, et al. The sustainability of energy conversion inhibition for tumor ferroptosis therapy and chemotherapy[J]. Small, 2021, 17(38):e2102695.
[35] Gao M, Yi J, Zhu J, et al. Role of mitochondria in ferroptosis[J]. Mol Cell, 2019, 73(2):354-363.e3.
[36] Chen Y, Qian J, Ding P, et al. Elevated SFXN2 limits mitochondrial autophagy and increases iron-mediated energy production to promote multiple myeloma cell proliferation[J]. Cell Death Dis, 2022, 13(9):822.
[37] Li C, Dong X, DuW, et al. LKB1-AMPK axis negatively regulates ferroptosis by inhibiting fatty acid synthesis[J]. Signal Transduct Target Ther, 2020, 5(1):187.
[38] Lee H, Zandkarimi F, Zhang Y, et al. Energy-stress-mediated AMPK activation inhibits ferroptosis[J]. Nat Cell Biol, 2020, 22(2):225-234.
[39] Dong S, Zheng L, Jiang T. Loss of lactate/proton monocarboxylate transporter 4 induces ferroptosis via the AMPK/ACC pathway and inhibition of autophagy on human bladder cancer 5637 cell line[J]. J Oncol, 2023, 2023:2830306.
[40] Dossou AS, Basu A. The emerging roles of mTORC1 in macromanaging autophagy[J]. Cancers (Basel), 2019, 11(10):1422.
[41] Yu X, Long YC. Crosstalk between cystine and glutathione is critical for the regulation of amino acid signaling pathways and ferroptosis[J]. Sci Rep, 2016, 6:30033.
[42] Conlon M, Poltorack CD, Forcina GC, et al. A compendium of kinetic modulatory profiles identifies ferroptosis regulators[J]. Nat Chem Biol, 2021, 17(6):665-674.
[43] Byun JK, Lee S, Kang GW, et al. Macropinocytosis is an alternative pathway of cysteine acquisition and mitigates sorafenib-induced ferroptosis in hepatocellular carcinoma[J]. J Exp Clin Cancer Res, 2022, 41(1):98.
[44] Armenta DA, Laqtom NN, Alchemy G, et al. Ferroptosis inhibition by lysosome-dependent catabolism of extracellular protein[J]. Cell Chem Biol, 2022, 29(11):1588-1600.e7.
[45] Zhang Y, Swanda RV, Nie L, et al. mTORC1 couples cyst(e)ine availability with GPX4 protein synthesis and ferroptosis regulation[J]. Nat Commun, 2021, 12(1):1589.
[46] Bhatt V, Lan T, Wang W, et al. Inhibition of autophagy and MEK promotes ferroptosis in Lkb1-deficient Kras-driven lung tumors[J]. Cell Death Dis, 2023, 14(1):61.
[47] Buccarelli M, Marconi M, Pacioni S, et al. Inhibition of autophagy increases susceptibility of glioblastoma stem cells to temozolomide by igniting ferroptosis[J]. Cell Death Dis, 2018, 9(8):841.
[48] Liu J, Liu Y, Wang Y, et al. TMEM164 is a new determinant of autophagy-dependent ferroptosis[J]. Autophagy, 2023, 19(3):945-956.
[49] Meyer N, Henkel L, Linder B, et al. Autophagy activation, lipotoxicity and lysosomal membrane permeabilization synergize to promote pimozide- and loperamide-induced glioma cell death[J]. Autophagy, 2021, 17(11):3424-3443.
[50] Zhang G, He J, Ye X, et al. β-Thujaplicin induces autophagic cell death, apoptosis, and cell cycle arrest through ROS-mediated Akt and p38/ERK MAPK signaling in human hepatocellular carcinoma[J]. Cell Death Dis, 2019, 10(4):255.
[51] Dasari SK, Bialik S, Levin-Zaidman S, et al. Signalome-wide RNAi screen identifies GBA1 as a positive mediator of autophagic cell death[J]. Cell Death Differ, 2017, 24(7):1288-1302.
[52] Lin Z, Niu Y, Wan A, et al. RNA m6A methylation regulates sorafenib resistance in liver cancer through FOXO3-mediated autophagy[J]. EMBO J, 2020, 39(12):e103181.
[53] He S, Li Q, Jiang X, et al. Design of small molecule autophagy modulators: a promising druggable strategy[J]. J Med Chem, 2018, 61(11):4656-4687.
[54] Galluzzi L, Green DR. Autophagy-independent functions of the autophagy machinery[J]. Cell, 2019, 177(7):1682-1699.
Lysosomal degradation systems in ferroptosis: quality control and energy metabolism
LIU Shizuo1, YANG Huan2, TANG Rong1, WANG Yanjiao3△
(1,,830017,;2,,830017,;3,,,,830017,)
Ferroptosis is a mode of cell death that is closely linked to autophagy. Autophagy, chaperone-mediated autophagy, and macropinocytosis are involved in the recycling of various intracellular biomolecules based on the lysosomal degradation system. In ferroptosis, autophagy-based catabolic processes selectively recognize ferroptosis-associated proteins and promote iron overload or lipid peroxidation. Moreover, autophagy or macropinocytosis inhibits ferroptosis by regulating the metabolic state of the cell or the degradation of extracellular proteins. Therefore, lysosomes play a "double-edged sword" role in ferroptosis. In this study, we describe the mechanisms of the lysosomal system in energy metabolism and the quality control of ferroptosis. We focus on autophagy and discuss the effects of different autophagy properties on ferroptosis. Then, the following review of the progress of ferroptosis research is provided.
ferroptosis; autophagy; lysosome
R329.21; R363.2
A
10.3969/j.issn.1000-4718.2023.09.019
1000-4718(2023)09-1691-06
2023-02-22
2023-06-11
新疆天山英才项目(No. 2021233);新疆医科大学博士启动基金(No. B202101)
Tel: 13659929362; E-mail: 13659929362@163.com
(责任编辑:卢萍,罗森)
