CRISPR 基因编辑技术在担子菌中的研究进展与应用
2023-10-10刘俊杰李辉平朱家漘李翠新曲绍轩
刘俊杰 李辉平 朱家漘,3 马 林 徐 平 李翠新 张 良 曲绍轩,3*
(1. 江苏省农业科学院蔬菜研究所 江苏省高效园艺作物遗传改良重点实验室,江苏 南京 210014;2. 西南林业大学生命科学学院,云南 昆明 650224;3. 江苏大学生命科学学院,江苏 镇江 212013;4. 江苏国耳生物科技有限公司,江苏盱眙 223001)
担子菌被视为一类菌丝体发达且可产生子实体的真核生物,其主要特征为具有担子的产孢结构。担子菌与人类的生产生活息息相关,在自然循环、经济循环、农业生态循环、大健康产业等方面都发挥重要作用[1]。大多数营养丰富的可栽培食药用菌都属于担子菌,如香菇(Lentinulaedodes)、双孢蘑菇(Agaricus bisporus)、糙皮侧耳(Pleurotusostreatus,俗称平菇)、灵芝(Ganodermalucidum)、猴头菇(Hericium erinaceus)等,是人类重要的膳食组成部分。其富含的一些天然活性成分,如菌多糖、灵芝酸,可增强人体免疫力,长期食用可降低患癌症的风险[2-3]。为充分利用其天然活性成分资源及加大对其生长发育的认识和改造,需要对靶基因进行精准的敲除、干扰或过表达等遗传操作。自然界中,担子菌、子囊菌以及不产生子实体的丝状真菌都是以菌丝体作为营养体进行生长,因此多利用菌丝体进行基因编辑;也有学者利用担孢子、孢子或子实体进行编辑[4]。但担子菌异核性、同源重组效率低、基因干扰脱靶率高、转化子遗传稳定性差等问题,严重阻碍了真菌遗传学的发展[5]。
核酸酶是裂解核苷酸磷酸二酯键的酶,可以是脱氧核糖核酸酶或核糖核酸酶、内切酶或外切酶、拓扑异构酶、重组酶、核酶或RNA 剪接酶[6]。细菌和古核细菌中特异性免疫响应的Cas 核酸酶在成簇规则间隔的短回文重复序列(CRISPR)指导下将入侵核苷酸进行切割沉默,为细菌和古细菌提供针对病毒和质粒的适应性免疫力,这种适应性免疫系统被称为CRISPR/Cas 系统[6-7]。2012 年,Jinek 等提出细菌的CRISPR/Cas 系统可应用于基因编辑[8],紧接着麻省理工学院的张锋团队在Science 发表论文,首次将CRISPR/Cas9 基因编辑技术改进并应用于哺乳动物和人类细胞,极大地推动了精准基因操作的进程[9]。CRISPR/Cas9 基因编辑技术首次应用于丝状真菌距今有十年多时间[10-11],大量的研究在一些子囊菌和担子菌等丝状真菌中成功实现基因编辑,然而效率却高低不一。本文主要回顾基因编辑技术在担子菌中的应用情况,围绕近年在基因编辑技术优化方面(启动子、密码子、供体形式、转化过程、受体形式、Cas蛋白等)的优化改进,为今后利用CRISPR/Cas 基因编辑技术满足人们对菌类作物产量、品质、抗逆性改良等方面的农业生产需求和营养保健的健康需求提供参考。
1 担子菌基因操作的发展
丝状真菌的基因操作奠基于1958 年,Emerson 通过采购的纤维素酶和蜗牛酶处理粗糙脉孢菌获得了形似原生质体的结构[12];次年,科学家Bachmann 和Bonner 使用相似的方法获得了高活性的粗糙脉孢菌原生质体[13],为进一步的基因操作带来可能。之后陆续发展的电转法导入质粒、农杆菌转化法(ATMT)、聚乙二醇介导法(PEG)等都基于此进行操作。Hinnen 等在酿酒酵母(Saccharomycescerevisiae)中利用PEG 介导的方式将leu2基因成功导入leu缺陷型酵母中,在此过程中,质粒成功整合到目标酵母的染色体组中,经验证在多个位点检测到leu2基因[14]。此后,其在丝状真菌中开始尝试,例如Kelly 等利用PEG介导的方式向黑曲霉(Aspergillusniger)中导入多个带有筛选标记的质粒,每微克获得大约4~20 个转化子[15];Michael 等尝试采用农杆菌转化法,成功将带有潮霉素(Hyg)抗性标记的Ti 质粒利用电激法导入到根癌农杆菌中,使其分别转染泡盛曲霉(A.awamori)、黑曲霉、镰孢霉(Fusariumvenenatum)、里氏木霉(Trichodermareesei)、粗糙脉胞菌(Neurosporacrassa)和双孢蘑菇的原生质体和分生孢子,皆获得了具有潮霉素抗性的后代[16-17]。
早先的基因操作依赖于细胞自身分裂期的同源重组现象,将外源基因整合到基因组中。该现象在自然状态下的发生率不到百万分之一,而在DNA 双键断裂(DSB)发生时进行基因操作概率可获得极大提升[18]。现代基因编辑技术主要着眼于向细胞内呈递人工生产的靶向核酸内切酶、经改造的核酸酶或能够表达该酶的质粒载体,来定点造成DNA 双键断裂。基于这一方向,锌指核酸酶(Zinc finger nucleases,ZFN)、转录活化因子样效应蛋白(Transcription activator-like effector,TALE)、类转录激活因子核酸酶(Transcription activator-like effector nuclease,TALEN)等技术先后问世,但都被CRISPR/Cas 基因编辑技术取代[6-7]。
2 CRISPR/Cas 系统基因编辑技术在担子菌中的应用现状
Cas 相关蛋白是一系列与CRISPR 簇临近的基因编码产物,主要参与核酸解聚、剪切、聚合等修饰过程,其中目前应用最为广泛的Cas9 是Ⅱ型CRISPR/Cas9 系统的特征蛋白,具有核酸内切酶的活性。CRISPR/Cas 的基因编辑系统在丝状真菌中进行遗传操作已形成一套基本的思路,即:先根据目的基因设计sgRNA、HDR(Homology direct repair)修复模板链、构建Cas 酶表达载体,然后制备真菌原生质体,再通过PEG 介导、电刺激等转化方式导入编码Cas 酶和sgRNA 的质粒或在体外制备的Cas 酶 + sgRNA复合体(RNP)。目前,基因编辑仍主要依靠DSB 出现所触发细胞自身的同源重组现象来引入外源基因,或经非同源末端连接NHEJ(Non-homology ending join)造成移码突变来敲除基因。而不同的修复途径将导致截然不同的基因编辑结果,HDR 途径会精确地敲入一段基因,使目的序列在可控范围内发生改变,且为分子标记的引入预留了充足的空间[19]。2013 年,Dicarlo 等首先将CRISPR/Cas9 基因编辑系统引入酿酒酵母,随后在构巢曲霉(A.nidulans)、粗糙脉孢菌、里氏木霉等多种丝状真菌中实现了基因编辑[20]。在此基础上,2016 年,宾夕法尼亚大学Yang Yinong 团队首先报道了通过CRISPR/Cas9 系统成功精准编辑双孢蘑菇多酚氧化酶(PPO)形成了抗褐变新品种,并获得商业化应用的许可——美国农业部(USDA)不会对使用CRISPR/Cas9 基因编辑工具进行基因改造的双孢蘑菇进行监管,这极大鼓舞了人们将CRISPR/Cas 系统应用在担子菌特别是食药用菌的新品种选育、定向改良、多基因功能利用、代谢途径重构等领域的研究[21]。
我国食药用菌CRISPR/Cas9 系统的建立主要借鉴于模式担子菌——灰盖鬼伞(Coprinopsiscinerea)遗传转化的成熟体系,最早开展基因编辑技术研究的是金针菇(Flammulinafiliformi)、灵芝和杏鲍菇(P.eryngii)[4]。华南农业大学林俊芳和郭丽琼教授团队2016 年最先尝试建立了金针菇CRISPR/Cas9 基因编辑体系(图1),该系统包含1 个Cas9 蛋白的表达载体、以金针菇的3-磷酸甘油醛脱氢酶基因Fvgpd为启动子驱动Cas9 蛋白,以潮霉素抗性基因(hph)为筛选标记,使用灰盖鬼伞上应用成熟的Ⅲ型启动子H1 作为sgRNA 的启动子构建基因敲除载体,通过PEG 介导的方法成功将靶标基因转化到金针菇原生质体内[22]。上海农业科学院尚晓冬研究员团队2017 年同样以Fvgpd为驱动Cas9 基因启动子和hph为筛选标记构建Cas9 表达载体,通过ATMT 转化法将靶基因导入金针菇单核菌株中获得了继代培养稳定的Cas9转化子,实现转化率6.84%,为金针菇建立高效的CRISPR/Cas9 基因编辑系统奠定了基础[23]。随后,华南农业大学林俊芳和郭丽琼教授团队在2018 年通过PEG 转化法获得了金针菇冷诱导相关的组氨酸激酶基因HK1 和HK2 的突变株,转化率分别为24.1%和12.5%[24];该团队以吲哚-3-甘油磷酸合酶基因(trpC)为驱动Cas9 基因启动子建立了隶属子囊菌亚门的蛹虫草(Cordycepsmilitaris)的CRISPR/Cas9 基因编辑系统,但未获得靶标基因ura3的突变株[25],直到2022 年蛹虫草的CRISPR/Cas 系统基因编辑技术才得到成熟应用[26]。南京师范大学陆玲教授团队2019 年对III 型启动子在杏鲍菇基因组中驱动sgRNA 转录的效率进行评估,筛选出4 个有功能的杏鲍菇内源U6 snRNA 启动子PEU6-1、PEU6-2、PEU6-3 和PEU6-4,其转录sgRNA 的活性强于体外T7 启动子的转录效率,最终获得了杏鲍菇尿嘧啶营养缺陷型突变株[27]。上海交通大学钟建江和肖晗教授团队2020 年利用灵芝内源u6启动子和有自我切割功能的核酶HDV 构建了一个有效的CRISPR/Cas9 系统,成功编辑了灵芝酸生物合成途径中重要的细胞色素P450 基因cyp5150l8,为抗癌次级代谢物灵芝酸的合成和调控机理的研究提供了重要平台[28-29]。继美国2016 年育出第一个基因编辑的抗褐变双孢蘑菇品种之后,国内在2020 年才报道在双孢蘑菇原生质体中成功建立了CRISPR/Cas9基因编辑系统,同时也对PPO 酶家族PPO4 基因进行多重位点突变,获得不同类型突变体组合以期获得不含有转基因成分的高效抗褐变双孢蘑菇新品种,但结果导致菌丝发育不良,证实了PPO4 可能主要参与双孢蘑菇的生长发育,而非抗褐变的关键基因[30]。

图1 担子菌门中的食药用菌CRISPR/Cas9 基因编辑技术的发展
2020 年后,CRISPR/Cas9 系统在担子菌上成功应用的报道才逐渐增多(图1),为深入开展担子菌基因功能研究和精准分子育种提供了无限的可能,尤其是构建出多基因敲除载体实现对基因组中多个靶位点进行编辑,为食药用菌品种的快速改良提供了新工具。Boontawon 等运用CRISPR/Cas9 构建的敲除体系分析了与平菇双核菌丝体形成相关的两个关键基因pcc1和clp1,揭示了clp1可能是参与形成锁状联合的关键基因[31];Xu 等选择平菇木质素降解相关的靶向基因vp1、vp2、vp3和62347,通过多顺反子tRNA和CRISPR 引导RNA 技术(PTG)构建一种载体表达多个sgRNA 的方法,开发出了平菇多基因突变的有效遗传工具[32]。
CRISPR/Cas 系统已作为第三代基因编辑技术在医疗、农业、生命科学基础研究等领域备受关注,但是与大多数非模式物种一样,该技术在食药用菌中的发展仍存在瓶颈,可供借鉴的稳定技术体系并不多[33]。虽然除Cas9 外,近年已有其他Cas 蛋白如Cas12a(cpf1)在丝状真菌中进行基因编辑应用[34],但在担子菌中至今未见报道。
3 担子菌CRISPR/Cas 系统的优化改进策略
使用CRISPR/Cas 系统进行基因操作需要先对Cas 酶供体方案进行选择并优化,不同的供体形式、供体类型都会影响基因编辑的效率,而不同的方案组合亦可适用于不同的应用场景。理论上,隶属于Ⅱ类的spCas9、dCas9、Cas12a、Cas13(用于编辑RNA)等核酸酶,在经人工设计的sgRNA 的引导下,可以应用于任何细胞基因的编辑,且编辑效率远高于上一代核酸酶。在细菌的基因操作中利用类别Ⅰ类C型Cascade-Cas3 的细菌自身CIRSPR/Cas 系统来进行多基因编辑,在铜绿假单孢菌中几乎实现100%的编辑效率[35]。因此针对不同细胞进行基因操作时,需根据具体情况进行适应性优化,才能实现快速高效的基因编辑效率。
3.1 Cas 酶的选择与优化
CRISPR/Cas 系统在实际应用中还是存在诸多限制,其中以脱靶效应和局限的PAM 位点(间隔序列向两端延伸的几个碱基都十分保守,通常由NGG 三个碱基构成)两方面最为突出,最初应用的spCas9就存在PAM 位点必须为“NGG”的限制。随着对CRISPR/Cas 家族研究的不断深入,Cas12、Cas13 等蛋白的发掘与应用极大地拓宽了应用范围[36]。
区别于Cas9,隶属于Ⅱ类V 型的cpf1(即Cas12a)与sgRNA 形成复合体时无需tracrRNA,仅通过单条42~44 个碱基的crRNA 便可以形成,因此其大小不到Cas9 的一半。更为重要的是,Cpf1 的切割位点位于PAM(T-rich)位点下游ts(target strain)序列23 bp,nts(none-target strain)序列18 bp 处,这就意味着其切割后断裂的区域天然地形成一个粘性末端,理论上来说这更加有利于带有同源臂的模版DNA与切口结引发HDR 修复途径[36]。CRISPR/Cpf1 系统在人类、小鼠细胞中的应用先例非常多,但在丝状真菌中的报道并不多。最早Vanegas 等在曲霉中构建Cpf1 载体成功敲除yA、albA两个基因,通过孢子颜色改变作为筛选标记[37];Kwon 等在嗜热假丝酵母(Thermothelomycesthermophilus)中分别使用来自弗朗西丝菌(Francisellanovicida)FnCpf1、氨基酸球菌(Acidaminococcussp.)AsCpf1 及酿脓链球菌(Streptococcuspyogenes)spCas9 三种Cas 酶通过RNP 导入的方法来编辑alp1、pks4.2、snc1、ptf14 个基因。结果显示,在pks4.2基因编辑组中,FnCpf1 和AsCpf1 的敲除效率分别是36%和10%,spCas9 可达到97%;在snc1基因编辑组中FnCpf1 与AsCpf1 的敲入效率达到52%和49%,而spCas9 为0;在alp1基因编辑组中FnCpf1和AsCpf1 的敲除效率达到91%~92%,而spCas9 仅有20%;在ptf1基因编辑组中,仅有spCas9 有20%的效率,Fncpf1为6%[38]。该研究揭示了不同的Cas 酶在不同的编辑方案中有着不同的效率,Cas9 高效作用于基因敲除,而在需要HDR 编辑的组别中却捉襟见肘,而两种Cpf1 在此时展现出了更高的编辑效率。同时,在编辑不同基因时效率也不尽相同。此外,Cpf1 在核黄菌(Ashbyagossypii)和棘孢曲霉(A.aculeatus)[39]中都有所报道,在多基因编辑和HR 途径诱导中表现出了更高的效率。因此合适的Cas 酶可以有效提升基因编辑效率,Cas9 系列更适用于基因敲除,而Cas12a 系列则更偏向于有HR 途径需求的基因编辑中。
以上两种是最基本的Cas 蛋白,不难发现局限的PAM 位点依然不可能适用于所有的场景。在Cas 蛋白结构中,PAM 序列主要位于NUC 结构域附近的WED、PI 结构域之间,而PAM 序列正是通过与PI 结构域中的氨基酸残基相互作用而起到特异性识别的作用。因此,通过对WED 与PI 结构域的改变,可以让Cas 蛋白识别并敲除任意感兴趣的位点。目前,数据库中已发表有超过1 000 种Cas9 蛋白序列[40],如:可高效编辑水稻基因,且识别位点为NGAN、NGCG 的SpCas9 VQR;用来纠正三倍致病性SNPs,识别位点为NG、NNG、GAA、GAT 和CAA 的xCas9-3.7 等,都结合实际情况采用定制化的Cas 蛋白实现了高效编辑的效果。
3.2 供体Cas 酶形式的选择优化
依托Cas 酶进行基因编辑依赖于其是否可在目的细胞内发挥作用。通过构建表达载体实现Cas 酶的体内表达是目前采用最为广泛的供体方案。在真菌细胞借助PEG 介导来进行编辑基因编辑时被广泛采用(表1)。Liu 等最先通过构建表达载体将CRIPR/Cas9 系统应用于里氏木霉,靶向敲除了其中的ura5基因,使其获得对5-FOA(氟乳清酸)的抗性,该靶标在多种真菌中被证实有效(野生型菌株中ura5的表达会将5-FOA 转化为具有细胞毒性的5-氟尿嘧啶)[57]。此后,CRISPR/Cas9 载体转化的方法相继被用于产黄青霉(Penicilliumchrysogenum)、烟曲霉(A.fumigatus)、卷枝毛霉(Mucorcircinelloides)等研究中[58-60]。
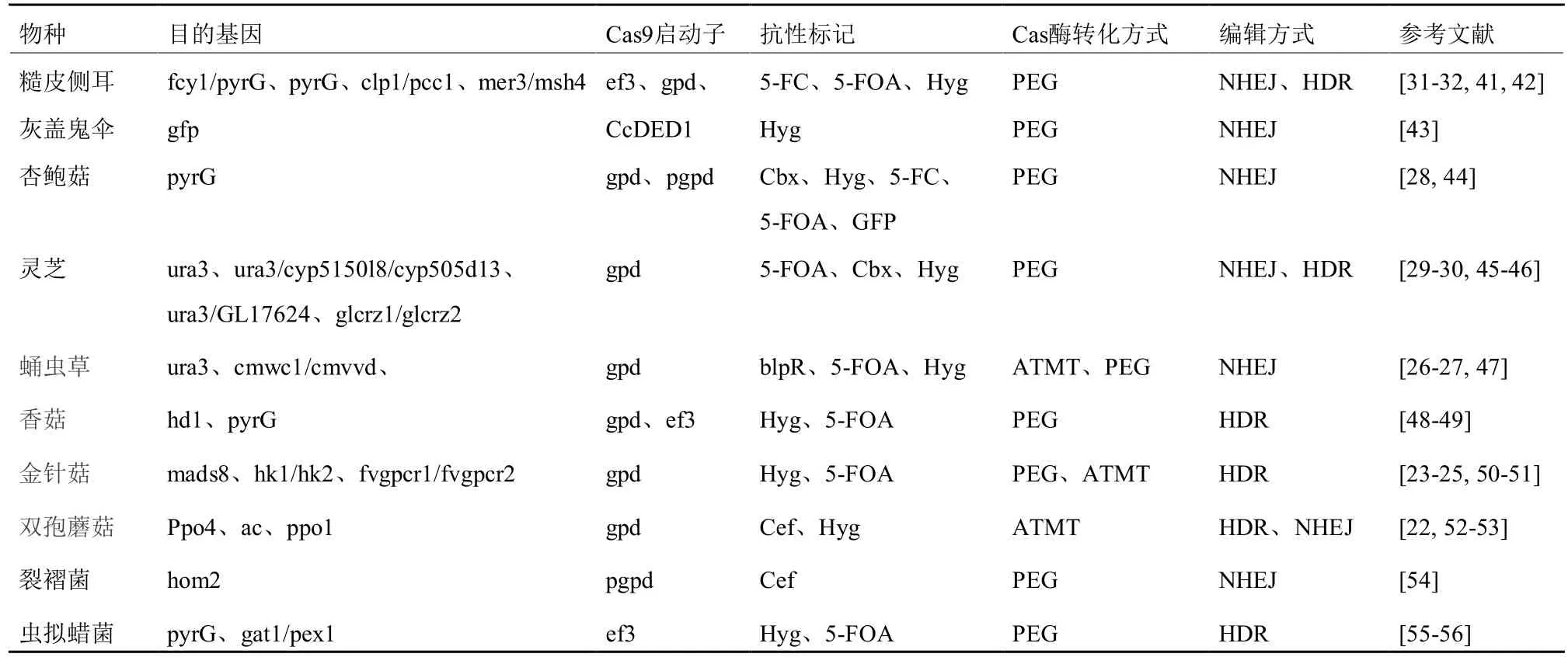
表1 担子菌CRISPR/Cas9表达载体优化策略和编辑效率
通过构建质粒表达载体对担子菌进行基因编辑时,需考虑以下方面:首先是密码子优化。异源表达对于靶细胞来说具有细胞毒性,因此在选择好合适的Cas 蛋白后,应根据靶细胞的密码子先进行同义替换来适应性地优化密码子;其次,载体应添加与靶细胞相适应的核定为信号(NLS,在真菌报道中SV40是常见的NLS[61])并选择合适的抗性标记,例如Cbx、Hyg、头孢霉素(Cef)等曾在真菌基因编辑中被报道[41,44];最后,选择合适的启动子来编码Cas 蛋白的转录翻译,tef1、gpdA、trpC启动子曾被报道于构巢曲霉中成功表达[62]。
利用表达载体实现Cas 酶的体内表达依然存在一些不可避免的风险:第一,存在同源序列的质粒有一定概率被整合到靶细胞基因组中,持续表达CRISPR/Cas 系统,在多次传代后,其不稳定遗传将造成难以预料的大规模脱靶效应;第二,多质粒呈递过程中,获得刚刚好的阳性转化子的概率并不高,因此多个途径的抗性筛选标记很重要;第三,某些稀有真菌可参考的基因数据有限,无法保证启动子的有效表达。同时,这一技术路线非常依赖目的细胞内的客观条件,启动子选择、密码子优化、抗性标记等客观条件都会影响基因编辑效率,从而大幅拉长试验周期。以上问题可采用RNP 作为另一条技术路线而有所改善。
Umeyama 等通过呈递RNP 的方式,在烟曲霉菌(A.fumigatus)中成功敲除了蛋白Cyp51A[63],Jan Vonk等在试管中合成并预先组装sgRNA+RNPs 复合体,以及修复模板链,通过PEG 介导的方式一起导入裂褶菌(Schizophyllumcommune)中实现了hom2基因的成功敲除[54];此外RNP 途径先后在镰刀菌(F.oxysporum)、灰霉菌(Botrytiscinerea)、里氏木霉(T.reesei)中都有所报道[64-66]。
RNP 策略存在编辑效率不稳定,在前述裂褶菌中甚至不到1%,因而在目前的基础上同样有很大优化空间。基于现有研究,推测造成RNP 编辑效率不稳定的原因主要是真菌原生质体的体积过小,限制了RNP 复合体的穿膜效率。Zou 等基于此,在对里氏木霉和蛹虫草的基因编辑中,于PEG 介导原生质体导入RNP 复合物的阶段,使用了0.006%的Triton X-100 孵育25 分钟增加细胞膜通透性,使用肌醇以大幅增加原生质体中单核细胞的比率,并辅以苯菌灵在加强单核细胞比率的同时使细胞周期同步化,最终在里氏木霉和蛹虫草中实现100%的编辑效率,其中在里氏木霉中HR 的发生率更是达到56.52%[67]。因此,在合适的优化手段与之匹配的情况下,RNP 可以作为更高效可靠的供体形式。
随着技术的改进,适用于RNP 的体外非细胞蛋白合成体系已相当成熟,相比传统的异源细胞表达体系来说,试验周期更短,获得的Cas 酶纯度更高。这些优势正将RNP 推向主流的供体形式。然而,拥有强大的工具并不意味着就可实现高效编辑,目的担子菌细胞的客观条件也不容忽视。
3.3 宿主受体优化
早先的研究发现,真菌强大的Ku 异源二聚体活性极易引导NHEJ 修复途径,很难触发基因断裂后的HDR 修复,从而造成试验预设以外的突变。真菌Ku70/Ku80 是一种能够与DNA 结合的异二聚体,可与DNA 依赖蛋白激酶(DNA-pkcs)、DNA 连接酶IVXRCC4 复合物和核酸外切酶形成多蛋白复合体,在DNA 发生双键断裂时快速滑向缺口,激活NHEJ 通路[68]。CRISPR/Cas 系统是否能躲避真菌宿主的免疫系统实现靶基因的编辑,是影响真菌高效基因编辑的又一重要因素。
Abdallah 等则通过RNP 的手段,在经ku80基因敲除改造的烟曲霉菌株中进行基因编辑,在同源臂长度为50 bp 的实验组中HR 发生率高达74%[69]。Ku70/Ku80 的敲除已在20 多个物种(主要为子囊菌)中证实,其被敲低或敲除后HR 的发生率大大提高(50%~100%)。并且一项平菇Ku70/Ku80 的敲除试验表明,经转化后的平菇样本菌丝活力、生长周期及子实体性状与野生型基本保持一致[70]。故在执行基因操作前先行制备Ku70/Ku80 等参与NHEJ 基因的敲除或沉默的工程菌株,几乎是高效率HDR 编辑的前提。
4 总结及展望
过去的基因编辑工具因成本高昂往往很少应用于丝状真菌,尤其是大部分非模式丝状真菌。CRISPR/Cas 系统相对较低的成本及优化门槛,给丝状真菌基因功能的研究带来了更多可能性。担子菌作为含丰富天然产物的大型真菌资源,其没有复杂的内含子作为干扰,更容易作为细胞工厂来生产人们所需要的物质。可产生子实体的大型食用真菌,不仅仅是健康膳食的重要组成部分,更有诸多证据证实其对人体健康的益处。此外,其在自然界中分解者的角色和易生长的特性,也不失为未来空间探索的优秀食源。故深入探究其内在分子机制及关键物质的代谢途径非常重要。
目前,在担子菌中广泛应用CRISPR/Cas 系统进行品种改良还存在诸多难点,有报道显示CRISPR/Cas的操作会对菌株生长发育产生负面影响。而担子菌中神秘基因簇是海量的,因而优化成本更低且可高通量多基因编辑的CRISPRa 和CRISPRi 未来可能更具优势[71-72]。
目前CRISPR/Cas 系统在担子菌代谢途径研究、工程菌优化、病原菌防治、遗传育种等多个领域显现出极大的潜力,在各菌种中的成功案例不断增多,可供借鉴的优化方案也会越来越多,这项技术在担子菌中广泛应用也势必拥有更为广阔的未来[53,72-73]。
