脲嘧啶除草剂氟嘧硫草酯的合成研究
2023-10-08刘安昌方强吴子豪徐婴兰陈典富
刘安昌,方强,吴子豪,徐婴兰,陈典富
(武汉工程大学化工与制药学院,武汉 430074)
20 世纪60 年代,美国杜邦公司研发上市了一系列脲嘧啶类除草剂,包括除草定、异草定、特草定和环草定等[1]。研究表明,这几种早期的脲嘧啶类除草剂作用于光合作用Ⅱ,是电子传递抑制剂。1986 年,瑞士Hoffman-La Roche 公司在对杜邦公司开发的早期脲嘧啶类除草剂进行优化时发现三氟甲基引入到脲嘧啶环能显著提高化合物的除草活性[2-4]。后续研究表明,在脲嘧啶环6 位上引入甲基苯基或并环时,化合物除草活性与三氟甲基类相近,相继开发出许多高效新品种,如苯嘧磺草胺、氟嘧硫草酯tiafenacil 等。氟嘧硫草酯作用于原卟啉原氧化酶,通过抑制原卟啉原氧化酶活性破坏植物体叶绿素的生物合成,导致有害物质原卟啉原IX 积累并渗透到细胞质,最终导致细胞膜破坏细胞死亡[5]。
氟嘧硫草酯由福阿姆韩农(FarmHannong)开发,CAS号:1220411-29-9;化学分子式:C19H18ClF4N3O5S;结构式如图1。

图1 氟嘧硫草酯结构式
根据文献报道,tiafenacil 的合成路线主要有如下2 条:
路线一:以三氟乙酰乙酸乙酯为原料,经过氨基化、环化、甲基化、磺化反应等8 步反应制备目标产物tiafenacil[6]。该合成路线使用了三氯氧磷和五氯化磷,导致后处理时产生大量酸性废水和磷化物。此外,采用昂贵的氯化亚锡还原苯磺酰氯制备苯硫酚,收率低,产生的三废较多,不适宜工业化生产。具体合成路线如图2 所示。

图2 氟嘧硫草酯合成路线一
路线二:以2-氟-4-氯苯胺为起始原料,经过酰胺化、环合、氯磺化、还原合成关键中间体3-(4-氯-2-氟-5-巯基苯基)-1-甲基-6-(三氟甲基)-2,4(1H,3H)-嘧啶二酮;以2-溴丙酸为原料经酸胺缩合制备中间体3-(2-溴丙酰氨基)丙酸甲酯;最后2 个中间体缩合方法同路线一,制备目标化合物tiafenacil[7-8]。具体合成路线如图3 所示。该合成路线使用的2-氟-4-氯苯胺比较昂贵,且不易购买。

图3 氟嘧硫草酯合成路线二
本文在合成路线二的基础上,经过改进,采用价廉易得的2-氟苯胺为起始原料,经过酰胺化、氯代、关环、甲基化、氯磺化、红磷还原、缩合等9 步反应制得目标物,具体合成路线如图4 所示。
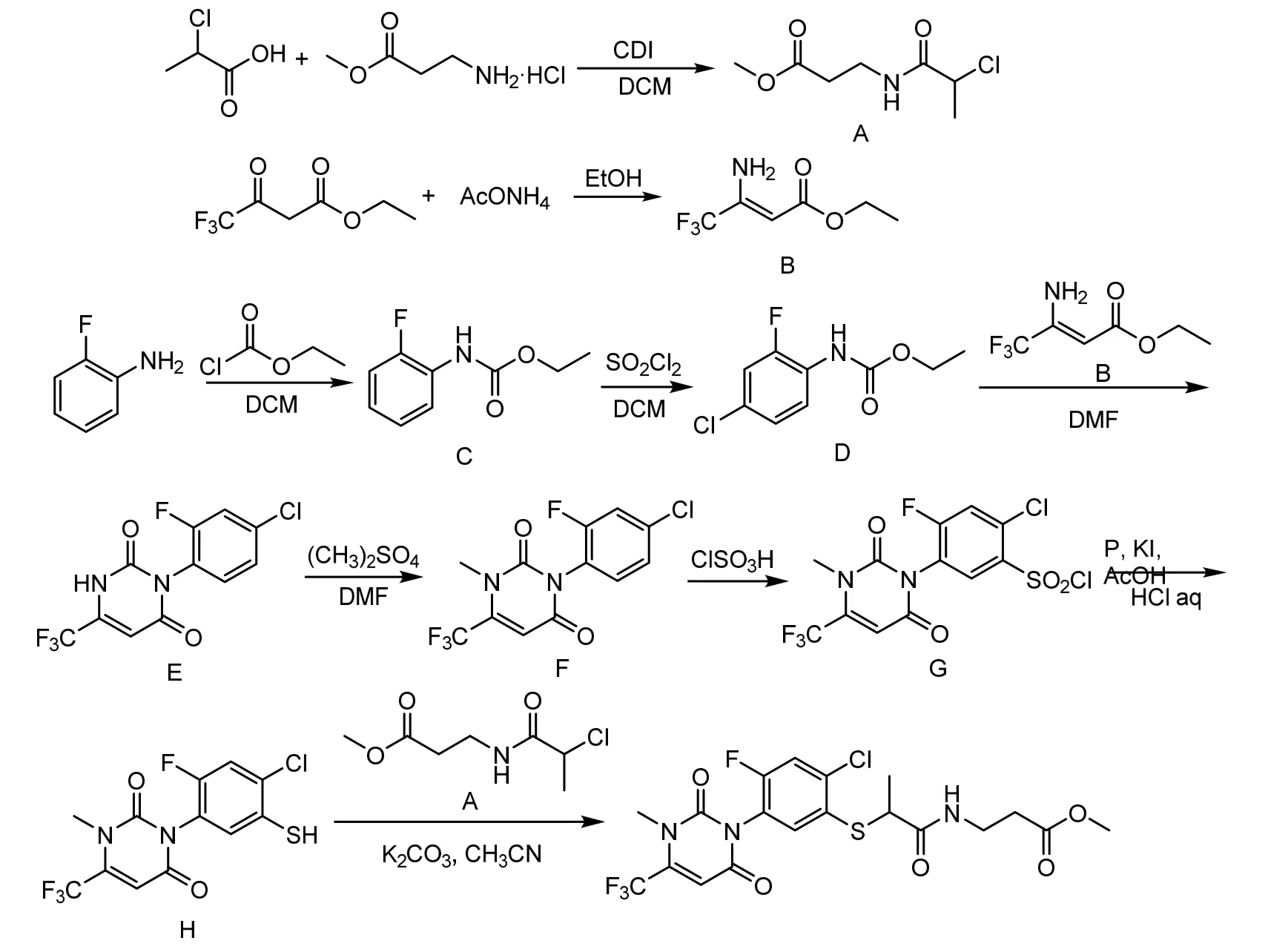
图4 氟嘧硫草酯的合成路线
1 实验部分
1.1 仪器与试剂
2-氟苯胺,上海麦克林生化科技有限公司;氯甲酸乙酯,武汉格奥化学技术有限公司;2-氯丙酸,前衍化学有限公司;3-氨基丙酸甲酯盐酸盐,泰坦科技有限公司;其他试剂均为市售化学纯或分析纯。1H 核磁谱用Bruker DPX-400 型超导核磁共振仪采集数据,以DMSO 或CDCl3为溶剂;熔点仪用WRS-3 数字熔点仪。
1.2 3-(2-氯丙酰氨基)丙酸甲酯(A)的合成
将16.3 g (0.15 mo1) 2-氯丙酸加入到120 mL二氯甲烷中,加入24.3 g(0.15 mo1)N,N'-羰基二咪唑,反应2 h 后,加入22 g(0.158 mo1)3-氨基丙酸甲酯盐酸盐,室温反应过夜。反应液依次用150 mL水、80 mL 10%(质量比)氢氧化钠、80 mL 5%盐酸洗涤,有机层用无水硫酸钠干燥、过滤、浓缩,得黄色透明油状物18.20 g,收率为62.9%。1H NMR(400 MHz, CDCl3)δ7.20 (s, 1H), 4.37 (q,J=7.0 Hz,1H),3.68(s,3H),3.52(q,J=6.1 Hz,2H),2.55(t,2H),1.68(d,J=7.0 Hz,3H)。
1.3 3-氨基-4,4,4-三氟丁烯酸乙酯(B)的合成
将92 g(0.5 mol)三氟乙酰乙酸乙酯、154 g(2 mol)醋酸铵和400 mL 无水乙醇加入到1 000 mL 的反应瓶中,升温至回流,反应10 h。停止反应,将反应液倒入250 mL 水中,用2×100 mL 二氯甲烷萃取,有机相依次用饱和碳酸氢钠溶液,食盐水洗涤,无水硫酸钠干燥,过滤,将浓缩得到的黄色透明液体减压蒸馏,收集40~46 ℃馏分无色液体79.10 g,收率为86.4%。1H NMR(400 MHz,CDCl3)δ6.19(s,2H),5.11(s,1H),4.16(q,2H),1.27(s,3H)。
1.4 N-(2-氟苯基)氨基甲酸乙酯(C)的合成
将33.3 g(0.3 mol)2-氟苯胺、35.55 g(0.45 mol)吡啶、150 mL 二氯甲烷加入装有搅拌器和温度计的圆底烧瓶中,在0 ℃下,滴加35.97 g(0.33 mol)氯甲酸乙酯,滴加完毕后升温至回流,继续反应2 h。冷却,过滤。滤液用2 mol/L 盐酸调节pH 至中性,然后用饱和食盐水洗涤,无水硫酸钠干燥、过滤,滤液浓缩得黄色透明油状液体50.75 g,收率为92.4%。
1.5 N-(4-氯-2-氟苯基)氨基甲酸乙酯(D)的合成
向装有36.6 g(0.2 mol)N-(2-氟苯基)氨基甲酸乙酯和150 mL 二氯甲烷的圆底烧瓶中缓慢滴加40.2 g(0.24 mol)磺酰氯,滴加完毕后室温反应过夜。加入100 mL 水,搅拌5 min,停止反应,分液。有机层依次用100 mL 水和100 mL 饱和食盐水洗涤。有机相用无水硫酸钠干燥、过滤,将滤液浓缩,得到白色固体白色晶体40.00 g,收率为92.2%。1H NMR (400 MHz, DMSO)δ9.39(s, 1H), 7.59 (t,J=8.7 Hz, 1H), 7.34 (dd,J=10.7, 2.3 Hz, 1H), 7.14 (d,J=8.7 Hz,1H),4.03(q,J=7.1 Hz,2H),1.16(t,3H)。
1.6 3-(4-氯-2-氟苯基)-6-(三氟甲基)嘧啶-2,4(1H,3H)-二酮(E)的合成
装有搅拌器,回流冷凝器的500 mL 四口烧瓶中,加入21.7 g(0.1 mo1)N-(4-氯-2-氟苯基)氨基甲酸乙酯、19.22 g(0.105 mo1)3-氨基-4,4,4-三氟巴豆酸乙酯和120 mLN,N-二甲基甲酰胺。然后将混合物搅拌升温至130~135 ℃,反应5 h。将反应液倒入300 mL冰水混合物中,用浓盐酸调节pH至1。用200 mL 乙酸乙酯萃取,有机相用3×100 mL 饱和氯化铵溶液洗涤。无水硫酸钠干燥,过滤。浓缩得米白色固体29.36 g,熔点为194~196 ℃,收率为95.3%。1H NMR (400 MHz, CDCl3)δ10.09 (s,1H),7.29(m,2H),7.21(dd,1H),6.25(s,1H)。
1.7 3-(4-氯-2-氟苯基)-1-甲基-6-(三氟甲基)-2,4(1H,3H)-嘧啶二酮(F)的合成
将15.4 g(0.05 mo1)3-(4-氯-2-氟苯基)-6-(三氟甲基)嘧啶-2,4(1H,3H)-二酮、13.8 g(0.1 mo1)无水碳酸钾和100 mLN,N-二甲基甲酰胺加入反应瓶中,在30 ℃下缓慢滴加13.86 g(0.11 mo1)硫酸二甲酯。滴加完毕后升温至60 ℃搅拌2 h。向反应混合物中加入100 mL 水,搅拌有固体颗粒析出。过滤,干燥得米白色固体15.70 g,熔点为78~80 ℃,收率为97.5%。1H NMR (400 MHz, CDCl3)δ7.28(m,2H),7.20(dd,1H),6.37(s,1H),3.56(s,1H)。
1.8 2-氯-5-[3,6-二氢-3-甲基-2,6-二氧-4-(三氟甲基)-1-(2H)嘧啶]-4-氟苯磺酰氯(G)的合成
将3-(4-氯-2-氟苯基)-1-甲基-6-(三氟甲基)-2,4(1H,3H)-嘧啶二酮16.13 g (0.05 mo1)和氯磺酸40.78 g(0.35 mo1)的混合物搅拌升温至150 ℃,反应4 h。冷却至室温,将反应液倒入200 mL 碎冰中并同时搅拌,过滤得灰粉色固体,用水洗至洗脱液呈透明无色,干燥,得灰白色固体15.90 g,收率为75.5%。1H NMR (400 MHz, CDCl3)δ8.13 (d,1H),7.57(d,1H),6.40(s,1H),3.58(s,1H)。
1.9 3-(4-氯-2-氟-5-巯基苯基)-1-甲基-6-(三氟甲基)-2,4(1H,3H)-嘧啶二酮(H)的合成
将乙酸(30 mL)、红磷5.43 g(0.175 mol)和适量的KI 混合搅拌并升温至80 ℃反应30 min 后,加入2-氯-5-[3,6-二氢-3-甲基-2,6-二氧-4-(三氟甲基)-1-(2H)嘧啶]-4-氟苯磺酰氯21 g (0.05 mol)的100 mL 乙酸溶液后,升温至110~115 ℃回流,通过薄层色谱TLC 检测反应进度。将反应液冷却过滤,将滤液移至反应瓶升温至100 ℃,滴加6 mL 1 mol/L HC1,滴加完毕升温至105~110 ℃,通过薄层色谱TLC 检测反应进度。停止加热,冷却后将反应液滴加到300 mL 水中,搅拌析出固体颗粒。过滤、水洗、干燥后得到灰白色固体13.00 g,收率为73.4%。1H NMR(400 MHz,CDCl3)δ7.46(d,1H),7.31(d,1H),6.36(s,1H),3.86(s,1H),3.55(s,1H)。
1.10 氟嘧硫草酯的合成
向3-(4-氯-2-氟-5-巯基苯基)-1-甲基-6-(三氟甲基)-2,4(1H,3H)-嘧啶二酮3.53 g(0.01 mol)和碳酸钾2.07 g(0.015 mol)中加入50 mL 甲苯,滴加3-(2-氯丙酰氨基)丙酸甲酯1.93 g(0.01 mol)和20 mL甲苯的混合物。滴加完毕后升温至70 ℃,搅拌3 h。停止反应,加入100 mL 分层,水相用少量甲苯。合并有机相,用饱和食盐水洗涤,用无水硫酸钠干燥、过滤,浓缩纯化得到3.58 g 固体,收率70.1%。1H NMR (400 MHz, CDCl3)δ7.45 (m, 1H),7.30(m,1H),7.15(m,1H),6.27(s,1H),4.10(q,1H),3.70 (s, 3H), 3.55 (m, 2H), 3.50 (s, 3H), 2.56 (t, 2H),1.69(d,3H)。
2 结论
本研究以2-氯丙酸为原料,3-氨基丙酸甲酯盐酸盐经酰胺化反应制备化合物(A)3-(2-氯丙酰氨基)丙酸甲酯;以三氟乙酰乙酸乙酯为原料,在乙醇中与醋酸铵共热合成化合物(B)3-氨基-4,4,4-三氟丁烯酸乙酯;以邻氟苯胺为原料,经酰胺化、氯代反应制备化合物(C)N-(4-氯-2-氟苯基)氨基甲酸乙酯;化合物(B)与化合物(C)在碱的作用下关环合成化合物(E) 3-(4-氯-2-氟苯基)-6-(三氟甲基)嘧啶-2,4(1H,3H)-二酮;化合物(E)经硫酸二甲酯甲基化,氯磺酸磺化,红磷/碘化钾/醋酸还原得到化合物(H)3-(4-氯-2-氟-5-巯基苯基)-1-甲基-6-(三氟甲基)-2,4(1H,3H)-嘧啶二酮;最后再与3-(2-氯丙酰氨基)丙酸甲酯缩合脱去一分子氯化氢得到目标化合物氟嘧硫草酯,收率70.1%。该合成路线工艺简单、原料易得,为工业化生产奠定了基础。
