青海草原毛虫转录组分析及SSR位点开发
2023-10-08南彦斌许嘉诚何啟玥潘学能周渊涛
南彦斌, 许嘉诚, 何啟玥, 潘学能, 周渊涛
(青海大学农牧学院, 青海 西宁 810016)
微卫星(Microsatellite)又名简单重复序列(Single sequence reperts,SSR),它由1~6个核苷酸碱基串联重复而形成,在真核生物基因组中分布比较广泛[1]。微卫星具有几个非常显著的特征,如可重复性高,共显性,多态性高,数量丰富,对基因组有较好的覆盖性等[2]。因此,其被广泛应用于群体遗传结构[3]、生物遗传多样性[4]、系统发生和遗传图谱绘制[5]等研究领域。虽然微卫星标记优点突出但是无法掩盖传统的SSR引物开发方法成本极高且成功率低的缺点[6]。目前,高通量测序技术和生物信息学的的快速发展,使得SSR标记开发更加高效便捷[7]。基于转录组数据已成功挖掘出多种昆虫的SSR位点。譬如李敏等[8]从中华大仰蝽(Notonectachinensis) 37 801条unigenes中搜索到3124个SSR位点,利用半定量实验筛选出16个SSR位点,他们认为SSR新标记的开发在中华大仰蝽的遗传多样性分析基因图谱构建中具有重要作用。王定锋等[9]基于转录组数据挖掘灰茶尺蠖(Ectropisgrisescens)微卫星标记,发现灰茶尺蠖转录组中SSR位点数量大、出现频率高、基元类型十分丰富,这些信息将为今后更好地利用SSR标记研究灰茶尺蠖种群间的遗传分化和制定灰茶尺蠖综合防治措施在分子生物学方面提供借鉴。韩小红[10]等通过对星天牛(Anoplophorachinensis)转录组SSR位点特征分析,发现其转录组中SSR的主要重复类型为单碱基重复,其次是三碱基重复。SSR位点信息和位点特征是昆虫分子标记研究的重要内容。然而截至目前,对于青海草原毛虫微卫星位点的研究还处于空白状态。
草原毛虫(Gynaephora)俗称草原毒蛾,隶属于鳞翅目毒蛾科。在世界范围内,目前有记载的草原毛虫属昆虫共有15种,而我国独占8种且大部分分布在青藏高原上,对青藏高原高寒甸草产生了严重的危害[11]。该虫通常一年发生一代,其1龄幼虫在土块、牛粪和草根下越冬,与国外的草原毛虫生活史周期相比国内的生活史周期较短[12-13]。由于青海草原毛虫对生境的要求比较特殊,故而在国内对它的研究较为有限,国外的相关研究也仅限于毒蛾科的其他虫种[14-15]。近十年从分子生物学的角度出发关于青海草原毛虫的研究,主要是从肠道菌、线粒体基因、序列变化和转录组基因表达等方面解析青海草原毛虫适应高海拔环境机制[15-18]。近年来有研究者从生物防治的角度开始探究青海草原毛虫的防治。如杨忠岐等研究金小蜂对青海草原毛虫的寄生作用[19],陈青红等研究4种生物药剂对青海草原毛虫的防控效果[20],白海涛等利用昆虫病原线虫防治青海草原毛虫21等。目前,随着国家对生态环境的重视程度的增加,未来借助于分子生物学研究,推动青海草原毛虫的生物防治是大势所趋。本研究通过高通量测序技术获得青海草原毛虫的转录组数据,结合生物信息学方法,对青海草原毛虫的SSR位点进行挖掘并进行序列特征的统计与分析,设计和筛选能够稳定扩增的SSR引物,为青海草原毛虫的分子鉴定与遗传多样性分析提供参考。
1 材料与方法
1.1 供试昆虫
本试验中的青海草原毛虫样品于2020年7月采自青海省海北州(36°59′ N,100°52′ E),设3个生物学重复,每组3头4龄幼虫及雌雄成虫,使用液氮快速冷冻后于—80℃超低温冰箱中保存。取5只单头4龄幼虫用于SSR引物验证,使用TIANamp Genomic DNA Kit基因组DNA试剂盒提取总DNA,经过2%的琼脂糖凝胶电泳和微量核酸分光光度计(北京凯奥科技发展有限公司)检测,结果满足进一步实验要求,置于-20℃保存备用。
1.2 总RNA提取、建库及数据评估
委托北京诺禾致源科技股份有限公司通过Trizol法提取青海草原毛虫样品RNA,经纯度和浓度检测合格后等物质的量混合,采用Illumina HiSeqTM2000高通量测序平台对青海草原毛虫雌雄成虫和4龄幼虫进行全长转录组测序、文库构建和数据评估。在得到原始数据后通过SMRTlink软件获得未组装的Subreads,进行校正,之后产生环形一致性序列(Circular-consensus sequence,CCS)。根据3′和5′引物及PloyA特征在CCS是否存在,从而确定全长序列和非全长序列,并分类找出全长非嵌合序列,通过同种类型聚类法(Iterative isoform-clustering,ICE)可将相似的全长非嵌合序列(Full length non-chimeric,FLNC)聚成一簇,得到Cluster,每个Cluster获得一条一致性序列,结合非全长序列对其进行校对(Polishing),筛选得到高质量序列用于后续分析[22]。
1.3 青海草原毛虫转录组unigenes的功能注释
在Trinity v2.0.2拼接的基础上,使用Corset v4.6软件进行转录本聚类,去除冗余转录本,得到非冗余unigenes。对聚类得到的unigenes使用blast2 go v2.5软件同GO,KOG和KEGG数据库比对,进行基因功能注释。
1.4 SSR的筛选
利用MISA软件(Micro satellite identification tool,www.pgrc.ipk-gatresleben.de/misa)对青海草原毛虫转录组中1 Kb以上的unigene进行SSR位点分析。对应的各个unitsize的最少重复次数分别为10,6,5,5,5和5,重复单元的长度大小分别是1,2,3,4,5和6 bp。两个相邻的微卫星重复单元间的间隔长度至少为100 bp。
1.5 SSR引物设计与验证
在1.4节筛选到的SSR位点的基础上,运用Primer3软件[23]对引物进行批量设计。目标扩增片段必须设置为包含SSR起始-3 bp,终止为+6 bp,扩增片段的长度为700~300 bp。引物的长度设为18~25 bp,最适长度为22 bp,引物最大能允许的不能识别的碱基数为1个,引物的最适退火温度为60℃,退火温度设置为55~65℃,上下游引物间的退火温度差异不能超过3℃,引物末端稳定性不能超过250[24]。
为了验证引物的稳定性,从批量设计的引物中随机选出25对引物,交由生工生物工程(上海)股份有限公司。PCR反应体系为25 μL:青海草原毛虫幼虫DNA模板1 μL,上下游引物(10 μmol·L-1)各加1 μL,2×EasyTaq®PCR SuperMix 12.5 μL,无核酸酶水9.5 μL。反应程序:94℃预变性5 min;94℃变性30 s,57℃退火30 s,72℃延伸40 s,总共35个循环;72℃延伸7 min,保存在4℃下。扩增后的产物用2%琼脂糖凝胶电泳检测(100V,30 min)。
2 结果与分析
2.1 青海草原毛虫转录组测序分析
采用Illumina HiSeqTM2000高通量测序平台对青海草原毛虫雌雄成虫、4龄幼虫转录组数据测序,获得71 038 419条原始reads;测序获得unigenes序列总长度为69 070 017 bp,碱基错误率为0.02%;Q20(质量值≥20碱基所占百分比)含量平均约为98.18%,Q30(质量值≥30碱基所占百分比)含量平均约为94.60%,测序质量较好。采用Trinity软件对clean reads数据进行拼接组装,组装后获得63 335条all unigenes。其中,N90(拼接转录本按照长度从长到短排序,累加转录本的长度,到不小于总长90%的拼接转录本的长度)和N50(拼接转录本按照长度从长到短排序,累加转录本的长度,到不小于总长50%的拼接转录本的长度)的长度分别为440 bp和1 714 bp,平均长度为1 091 bp。所有63 335条unigenes序列用于后续的SSR搜索。
2.2 青海草原毛虫转录组的GO注释
利用Blast2GO v2.5进行GO注释,将注释成功的基因按照GO三个大类(生物学过程,细胞组成,分子功能)的下一层级进行分类。结果表明(图1),18 588条unigenes共得到78 461条功能注释,分布在43个功能区。其中,获得注释序列数最多的是生物学过程分类,有39474条,分布在26个功能区;其次是分子功能分类的注释序列,有21 443条,分布在12个功能区;注释序列最少的是细胞组分类别,为17 544条,分布在5个功能区。在生物学过程类别中,细胞进程和代谢进程的注释序列数占主导地位,分别为10 846和9 355条。在分子功能分类中,注释到结合和催化活性的序列数最多,分别为9 932和7 295条。在细胞组分分类中,注释到细胞结构体和胞内部分的序列占主导地位,分别为8 267和4 877条。有11个功能区的注释序列超过2 000条,分别是细胞进程(10 846),结合(9 932),代谢过程(9 355),细胞结构体(8 267),催化活性(7 295),生物调节(4 085),生物调控进程(3 803),含蛋白复合物(3 490),定位(3 002)和应激响应(2 607)。
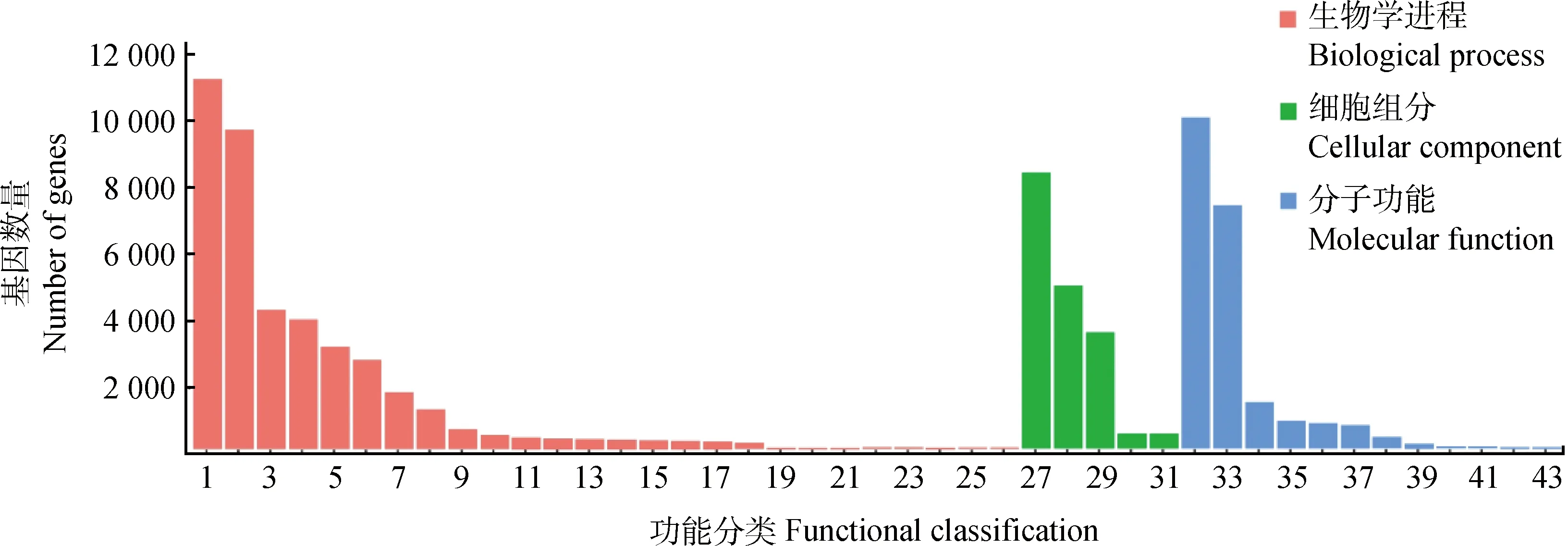
图1 青海草原毛虫unigenes GO功能注释
2.3 青海草原毛虫Unigenes的KEGG分类
KEGG代谢通路富集分析结果表明(图2),共有11 846条unigenes参与到细胞进程、环境信息处理、遗传信息处理、新陈代谢和有机体系统这五大类生化代谢通路中。其中代谢系统途径中基因最多(3 346条),它们主要参与碳水化合物代谢和脂质代谢等过程,分别为566条和436条。在这34组代谢通路子类别中,信号转导(Signal transduction)通路的基因最多,为1 302条。这些代谢通路相关的基因分布于已知的284个代谢通路,其中富集最多的10条通路分别是核糖体,内质网蛋白加工,嘌呤代谢,RNA转运,剪接体,氧化磷酸化,碳代谢,嘧啶代谢,cAMP信号通路,真核生物的核糖体生物发生,真核生物中的核糖体生物合成(表1)。

表1 KEGG数据库中单基因的十大代谢途径富集
2.4 青海草原毛虫Unigenes的KOG分类
对有注释的基因进行直系同源分类(图3),9 521条unigenes共得到10 628条注释,分为26类,其中,一般功能预测和翻译后修饰的基因最多,分别为1 487条(13.99%) 和1 114条(10.48%);其余依次是T类信号转导机制,有1 005条(9.46%);J类翻译、核糖体结构和生物合成,有864条(8.13%);K类转录,有644条(6.06%);U类细胞内运输、分泌和囊泡转运,有639条(6.01%);S类功能未知,有630条(5.93%);A类核酸处理与修饰,有598条(5.63%)。其余的都低于500条,X类未命名蛋白质,有1条(0.01%)。

图3 青海草原毛虫unigenes KOG分类注释
2.5 青海草原毛虫转录组中SSR分布特征
对青海草原毛虫转录组中的63 335条unigenes的数据利用MISA软件进行简单重复序列(SSR)位点搜索。总共找到12 597个微卫星位点,分布于9 851条unigenes中,其微卫星出现频率(含有SSR的unigenes数量与总unigenes数量之比)是15.55%,其中含有1个以上SSR位点的独立基因序列有1 852条,以复合型形式存在的SSR有801个,SSR的分布频率(SSR个数与总unigene数量之比)为19.89%(表2)。这些SSR基序包含1~5 bp的串联重复序列。

表2 青海草原毛虫转录组SSR分布及特征
2.6 青海草原毛虫转录组序列中SSR位点重复类型分析
从青海草原毛虫转录组数据中可以得到,单核苷酸重复是最主要的重复,占SSR总数的71.83%。二核苷酸重复居其次,占SSR总数的14.27%。三、四核苷酸重复,分别占SSR总数的10.59%和2.99%。五核苷酸和六核苷酸重复相较于前几种核苷酸的含量很少,分别占SSR总数的0.29%和0.0.3%。出现频率最高的是10次重复基元,共5 265个SSR位点,占总SSR位点的41.77%(表3)。
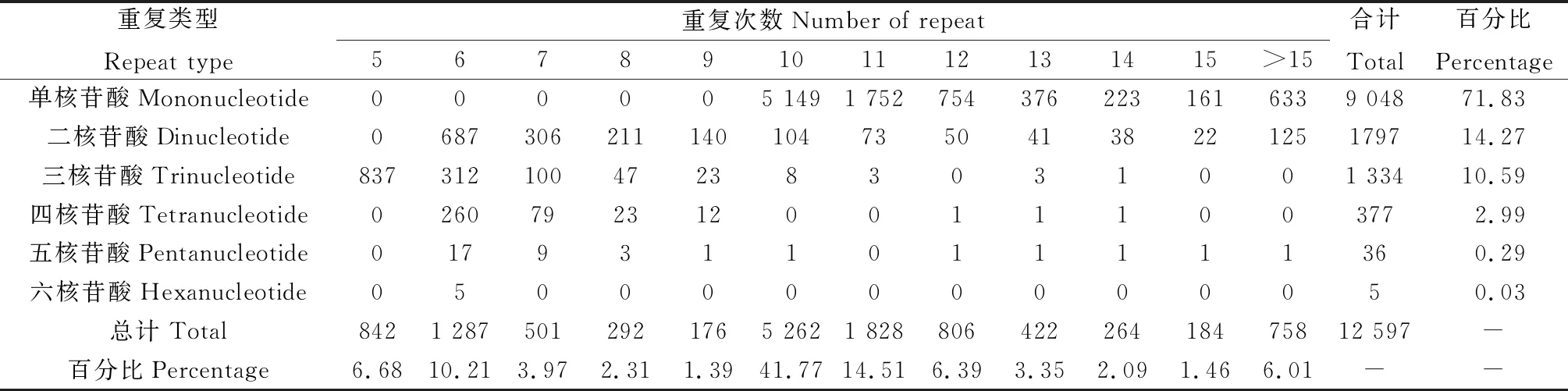
表3 青海草原毛虫转录组数据的SSR重复类型分布
对5~99次SSR基元重复次数进行分析结果表明(图4),SSR位点重复在5~10次的有8 335个,占总SSR位点的66.17%;重复次数在11~20次的有3 896个,占总SSR位点的30.93%;重复次数在21~30次的有267个,占总SSR位点的2.12%;重复次数在31~40次的有53个,占总SSR位点的0.42%;重复次数在41~50次的有20个,占总SSR位点的0.16%;重复次数在51~99次的有26个,占总SSR位点的0.21%。对单核苷酸重复,二核苷酸重复和三核苷酸重复的分析发现,SSR的数量与重复次数成反比出现,随着重复次数的增加,SSR的数量反而在减少。在图中未呈现的四、五、六核苷酸重复具有与单、二、三核苷酸重复相同的趋势表现。

图4 青海草原毛虫转录组中SSR基元重复次数
2.7 青海草原毛虫转录组基元类型分析
青海草原毛虫转录组SSR基元类型结果显示(图5和表4),有60个SSR重复基元类型,共获得12 597个SSR位点。其中,2种类型的基元在单核苷酸重复中呈现,(A/T)n是最主要的基元类型,形成8990个SSR位点,占同类基元SSR位点的99.36%,占总SSR位点的71.37%。4种类型的基元在二核苷酸重复中呈现,(AC/GT)n为主要优势基元,形成763个SSR位点(42.46%,6.06%),其次为(AT/AT)n和(AG/CT)n基元,分别形成619个(34.45%,4.91%)和370个SSR位点(20.59%,2.94%)。10种类型的基元在三核苷酸重复中呈现,(AAT/ATT)n是最主要的优势基元,有533个SSR位点(39.96%,4.23%);其次为(ATC/ATG)n,(AAC/GTT)n,(AAG/CTT)n。分别形成236个(17.69%,1.87%),142个(10.64%,1.13%),114个(8.55%,0.91%)SSR位点。16种类型的基元在四核苷酸重复中呈现,(AAAT/ATTT)n是最主要的优势基元,有170个SSR位点(45.09%,1.35%);其他优势基元分别为(ACAT/ATGT)n、(AGAT/ATCT)n,分别形成57个(15.12%,0.45%)和44个(11.67%,0.35%)SSR位点。在五核苷酸重复中有22个重复基元类型,最主要的优势基元是(AAAAT/ATTTT)n,(AACGT/ACGTT)n,在这两个基元中都形成了4个(11.11%,0.03%)SSR位点。六核苷酸重复有5个重复基元类型,全都形成1个SSR位点(20%,0.01%)。

表4 青海草原毛虫转录组不同重复基元SSR位点数及频率

图5 青海草原毛虫转录组不同重复类型优势基元SSR位点数分布
2.8 青海草原毛虫SSRs引物验证
本研究从被筛选出的微卫星引物中随机选取25对,对5个不同样品(海北州同一地点的5个青海草原毛虫个体重复)的青海草原毛虫DNA样本进行PCR扩增,结果11对引物在每个样品中扩增取得的片段与预期的片段几乎一致。有效扩增率为44%(表5,图6)。

表5 青海草原毛虫部分EST-SSR筛选引物信息

图6 青海草原毛虫5个样品25个SSR位点引物的PCR扩增产物电泳结果
3 讨论
伴随着高通量测序技术的快速发展,对昆虫转录组学的研究变得非常广泛[25]。本研究测序获得unigenes序列总长度为69 070 017 bp,平均长度为1 091 bp,N50长度为1 714 bp,N90长度为440 bp。由于N50的值较大,因此其组装完整性较高,保证了转录组分析的准确性。
本研究发现,青海草原毛虫转录组数据库中存在大量的微卫星,且种类丰富,核苷酸的重复类型出现了6种。单核苷酸重复中最主要的重复类型是A/T,这与对印度谷螟(Plodiainterpunctella)[2]、黏虫(Mythimnaseparata)[26]、褐飞虱(Nilaparuatalugens)[27]SSR位点特征的研究结果基本相同。但本次试验结果与对黄粉虫(Tenebriomolitor)[28]、麦红吸浆虫(Sitodiplosismosellana)[29]、沙葱叶甲(Galerucadaurica)[30]等昆虫的SSR位点特征研究结果相比较存在差异。这些研究发现核苷酸的重复类型以单核苷酸为主,三核苷酸次之。本研究的结果是单核苷酸重复是最主要的重复类型,二核苷酸次之。该结果与意大利微蝗(Calliptamusitalicus)[31]、梨小食心虫(Grapholithamolesta)[32]、沙棘木蠹蛾(Holcocerushippophaecolus)的研究结果类似。通常情况下,在ESTs里,单核苷酸为主要重复类型,三核苷酸重复居于其次。原因在于,与编码区其他的重复基元类型相比,三核苷酸核心基元更加稳定,基本不会出现编码框滑动突变现象[33]。本研究中出现差异,可能是不同样品之间本身存在差异,从而导致结果不同。
为验证青海草原毛虫SSR的稳定性,本研究利用其转录组数据设计出2 714对引物,从中随机选出25对引物进行扩增试验,结果表明11对引物在每个单个样品中都能成功扩增,并得到预期条带。这些位点的多态性目前还不明确,需要进一步试验评估,但它的数量足以开展青海草原毛虫种群遗传学相关研究。导致不同样品的EST-SSR位点扩增失败的原因有很多,如:由于基因组DNA中的内含子在转录后没表现出来,对扩增过程造成干扰从而导致预期的条带与实际扩增的条带相比,会出现扩增条带远远大于预期的现象;在基因组中EST-SSR的表达丰富度太低,导致产物的浓度低,无法检测[2]。
微卫星的重要作用体现在基因调控、蛋白功能、种群遗传结构、种群扩散及迁移等方面[34]。引物的开发是微卫星应用的前提,传统的方法开发成本太高,不合时宜。通过在转录组高通量数据中直接发掘微卫星并设计引物的方法,大大减少了开发成本,很大程度上对微卫星的开发进程起到推动作用[30]。本研究在青海草原毛虫转录组数据库中共筛出12 597个SSR位点,并对其分布情况及可用性进行了详细叙述,这些SSR的发掘,对于之后研究青海草原毛虫的遗传多样性和功能基因组学奠定了坚实的基础,也更进一步表明通过高通量测序技术发掘昆虫的SSR引物是一种符合人类需求的研究方法。
4 结论
本研究通过KOG,GO注释和KEGG通路三大数据库分析,发现unigenes注释主要集中于一般功能预测、细胞进程和碳水化合物代谢。本研究共筛出12 597个SSR位点,发生频率为19.89%,SSR位点数量丰富,(A/T)n,(AC/GT)n分别是单核苷酸重复和二核苷酸的优势基元,SSR数量随重复次数增加而降低,重复次数类型随基元序列长度增长而减少。从1 556条青海草原毛虫unigenes中成功设计出2 714对青海草原毛虫SSR引物,随机挑出25对引物进行PCR验证,有11对引物扩增出目的DNA片段。本研究基于转录组数据成功筛选出青海草原毛虫微卫星位点,获得的青海草原毛虫SSR位点为进一步研究其种群遗传学和种群扩散及迁移奠定基础。
