基于临床验方的六金颗粒制备及质量标准研究
2023-09-28罗毅秦金淼张荣飞刘子夜曹桂云姚元成孟兆青容蓉
罗毅, 秦金淼, 张荣飞, 刘子夜,曹桂云, 姚元成, 孟兆青, 容蓉
1. 山东中医药大学 药学院,济南 250355;2. 山东省中医药治疗呼吸系统疾病技术创新中心,济南 250103;3. 山东宏济堂制药集团股份有限公司,济南 250103
六金颗粒由蒲公英、 忍冬藤、 大青叶、 板蓝根、 锦灯笼、 龙脷叶6味药材组成, 具有清热解毒、 疏风利咽的功效. 方中蒲公英甘寒, 擅疏风清肺热, 为君药; 忍冬藤苦甘寒, 助君药清热解毒, 能利尿通淋, 使热从小便而出, 兼有消肿散结、 利咽化痰之功效, 为臣药; 大青叶、 板蓝根清上焦热, 尤善清血中之热, 且能利咽消斑; 锦灯笼清热利尿通淋; 龙脷叶甘淡润肺, 避免热伤肺阴, 同时兼有通便作用, 使热从大便而出, 共为佐使. 六药合用, 专于清肺热, 兼清上中下三焦之热, 能清气分、 血分之热, 使热从内消、 从二便而解. 本方专于清热以治病之本, 同时疏风、 利咽消肿、 止咳化痰以解症之标, 且清热兼顾护阴, 使邪去而不伤正. 基于该处方良好的临床应用实践, 可进行中药新药的开发. 本研究对该方剂制备工艺反复优化确定其参数并对制剂进行质量标准研究.
君药蒲公英为菊科植物蒲公英、 碱地蒲公英或同属数种植物的干燥全草, 含有多种成分, 可分为三萜类、 黄酮类、 倍半萜内酯类、 植物甾醇类、 香豆素类、 色素类、 酚酸类、 脂肪酸、 挥发油、 胡萝卜素类等. 临床药理研究蒲公英具有显著的广谱抑菌、 抗病毒及免疫调节等作用[1-2], 与六金颗粒功能主治相关. 通过前期对六金颗粒制剂物质基础分析, 菊苣酸为蒲公英中的专属性成分, 在制剂中与其他化合物相比含量较高. 菊苣酸是一种羟基肉桂酸, 分子式为C22H18O12, 现有文献报道证明其对呼吸道合胞病毒、 单纯疱疹病毒等多种病毒以及金黄色葡萄球菌、 结核分歧杆菌等不同致病细菌具有显著的抑制活性[3-4]. 本研究选择与制剂药效相关的活性成分作为定量评价六金颗粒质量的指标.
1 试验方法
1.1 仪器与药材
1.1.1 仪器
试验所用仪器主要有: Aglient 1260高效液相色谱仪(安捷伦科技有限公司), BSA224S-CW电子天平(赛多利斯科学仪器(北京)有限公司), XS-105电子天平(梅特勒托利多科技(中国)有限公司), LPG-8精密药用喷雾干燥机(山东精诚医药装备制造有限公司), GFL25P干法制粒机(山东精诚医药装备制造有限公司)以及KQ800VSM双频静音型超声波清洗器(昆山市超声仪器有限公司).
1.1.2 药材
蒲公英(购自洛宁益民药材药种购销站, 批号: YF22100012)、 忍冬藤(购自河北一仁药业有限公司, 批号: 21030011)、 大青叶(购自安国市世元商贸有限公司, 批号: YF21040002)、 板蓝根(购自山东兴泰中药材有限公司, 批号: 21090014)、 锦灯笼(购自河北一仁药业有限公司, 批号: YF21030001)、 龙脷叶(购自河北一仁药业有限公司, 批号: YF21030003)等药材经山东省食品药品检验研究院林永强鉴定, 均符合《中华人民共和国药典》2020年版一部规定.
麦芽糊精(购自长岭吉隆生物药业有限公司, 批号: 2010001)、 甜菊糖苷(购自曲阜圣仁制药有限公司, 批号: 22072102)、 菊苣酸对照品(纯度: 99.1%, 批号: 11752-202104, 购自中国食品药品检定研究院)、 酸浆苦味素L对照品(供薄层鉴别用, 批号: 111831-201903, 购自中国食品药品检定研究院); 乙腈和甲醇为色谱级, 均购自月旭科技(上海)股份有限公司; 无水乙醇购自天津市富宇精细化工有限公司; 磷酸购自天津市科密欧化学试剂有限公司.
1.2 方法
1.2.1 六金颗粒的制备
蒲公英、 忍冬藤、 板蓝根、 大青叶各600 g, 锦灯笼、 龙脷叶各150 g, 加水煎煮2次, 第1次1 h, 第2次0.5 h, 合并煎液, 滤过, 滤液浓缩至相对密度1.09~1.11(60 ℃), 滤过, 喷雾干燥, 喷干粉过100目筛, 加入甜菊糖苷和麦芽糊精适量, 制粒, 制成1 000 g, 即得.
1.2.2 颗粒评价指标的测定方法
采用成型率、 堆密度和休止角3个指标来评价颗粒的质量, 以及颗粒吸湿性和水分含量, 以上5个指标综合评价辅料的选择以及制剂的成型工艺[5-8].
成型率测定方法. 将六金颗粒称质量, 先过1号筛, 再过5号筛, 能过1号筛但不能通过5号筛的颗粒作为合格颗粒, 收集并称质量, 颗粒的成型率=(合格颗粒质量/过筛前颗粒质量)×100%.
堆密度测定方法. 采用量筒测定法, 称取制得的颗粒10 g, 放入干燥的25 mL量筒中, 记录其体积(mL), 堆密度值(g/mL)=10/c. 重复测定5次, 取平均值.
休止角测定方法. 采用固定漏斗法, 3只漏斗串联后, 将其固定于水平放置的坐标纸上高1.5 cm的位置(H), 将六金颗粒沿漏斗壁缓慢地倒入最上面的漏斗中, 直到坐标纸上形成的颗粒圆锥尖端可以接触到漏斗口, 测算颗粒底部的直径(2R), 计算休止角tgα=H/R. 重复测定3次, 取平均值.
吸湿性测定方法. 精密称定本品颗粒2 g, 平行3份, 置于干燥器中25 ℃恒温平衡48 h, 将底部放置氯化钠过饱和溶液, 此时干燥器内相对湿度为75%. 于24 h, 48 h, 72 h, 96 h, 120 h, 144 h, 168 h后称质量, 计算吸湿百分率. 吸湿百分率=(吸湿后颗粒质量-吸湿前颗粒质量)/吸湿前颗粒质量×100%. 吸湿二项式方程:W=2at+bt+c, 其中W表示吸湿量,t表示时间.
水分测定方法. 取制得的颗粒2~5 g, 各3份, 采用烘干法测定.
2 结果与分析
2.1 处方中辅料的筛选
2.1.1 喷干粉与辅料比例考察
称取喷干粉平均分为5份, 分别按照药辅比1∶0.25加入相应辅料, 混合均匀, 制粒. 以过筛情况、 颗粒成型率、 流动性及溶化性为考察指标, 优选最适合的辅料, 结果见表1.
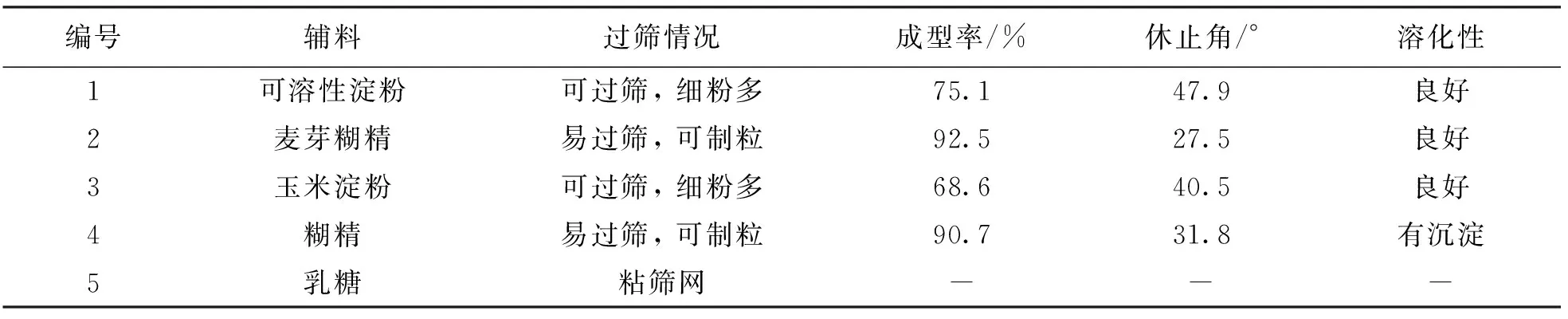
表1 辅料种类筛选
由表1可知, 在成型工艺中, 除乳糖外, 其余4种辅料均可制粒, 但可溶性淀粉和玉米淀粉组制粒过筛时细粉较多, 成型率低于糊精和麦芽糊精组; 以麦芽糊精制粒成型率和流动性均优于以糊精制粒, 并且以糊精制粒后用热水溶解, 有明显白色沉淀, 因此本品选择麦芽糊精为辅料进行制粒.
称取喷干粉平均分成3份, 分别加入不同量的麦芽糊精, 混合均匀, 制粒. 以过筛情况、 颗粒成型率、 流动性及颗粒水分为考察指标, 优选麦芽糊精的用量, 结果见表2.

表2 辅料用量筛选
由表2可知, 成型工艺中, 3个药辅比过筛情况一致, 均为已过筛可制粒; 药辅比为1∶0.25和1∶0.30的成型率及流动性优于1∶0.20, 并且药辅比为1∶0.25和1∶0.30颗粒水分含量低于1∶0.20; 药辅比1∶0.25和1∶0.30两组间的成型率、 流动性及颗粒水分含量差异小, 因此基于辅料添加最小量原则, 选择喷干粉∶麦芽糊精比例为1∶0.25作为制粒的辅料添加量.
2.1.2 矫味剂的筛选
为对颗粒进行矫味, 针对三氯蔗糖、 甜菊糖苷和阿斯巴坦3种矫味剂种类及用量进行了口感打分评价, 由50名健康实验员进行打分, 结果表明, 添加5‰甜菊糖苷作为矫味剂的颗粒评分最高, 因此选用5‰甜菊糖苷作为矫味剂(表3).
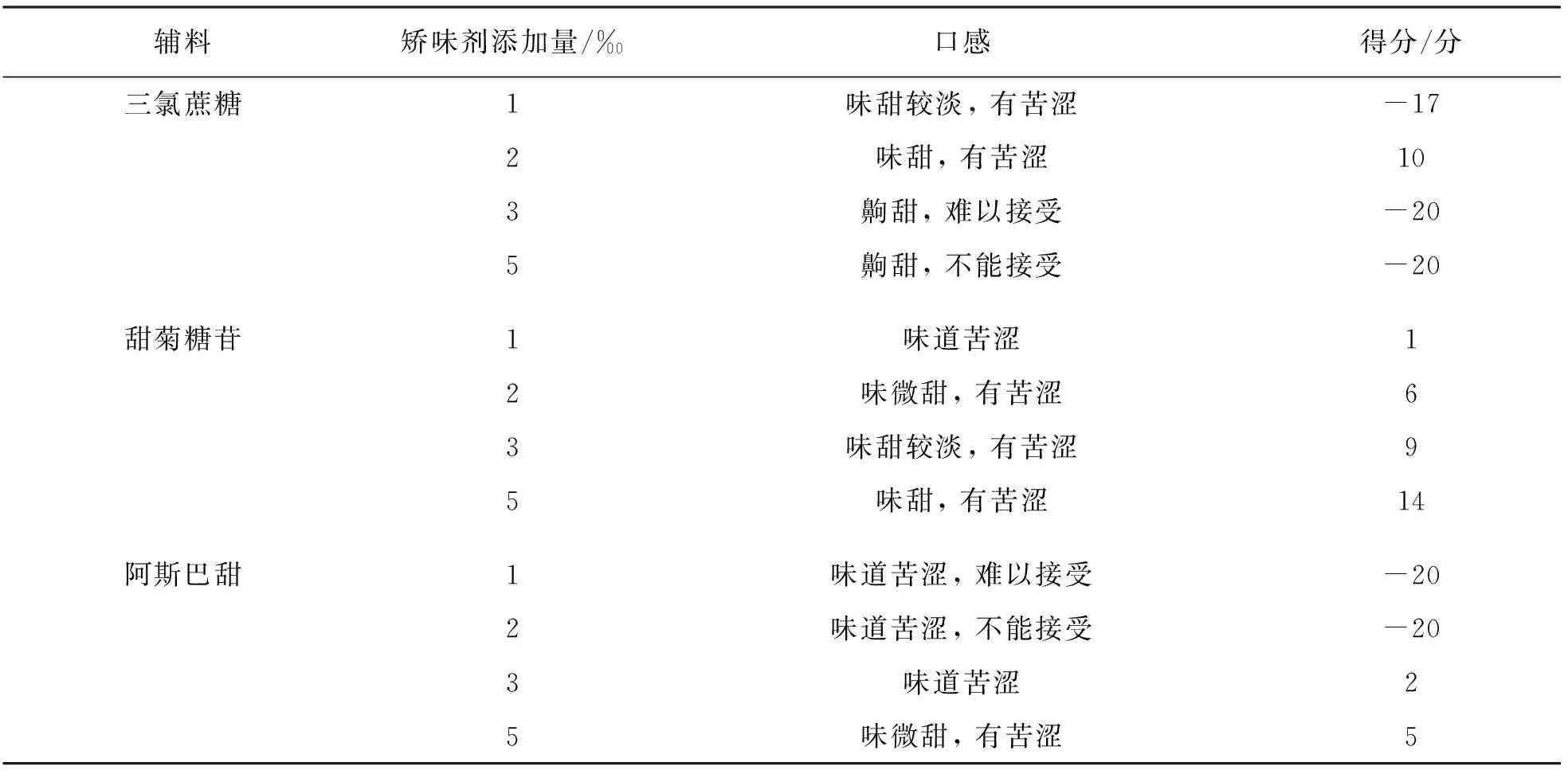
表3 不同矫味剂及用量
2.1.3 中试工艺验证
连续3批六金颗粒中试生产, 采用干法制粒, 根据药辅比1∶0.25加入麦芽糊精, 加入5‰甜菊糖苷作为矫味剂, 制粒后分单包装. 按照《中国药典》2020年版四部颗粒剂相关检查项对其进行检查. 3批次六金颗粒粒度均匀, 颗粒颜色由黄棕色至深棕色; 按照要求, 能通过一号筛但不能通过五号筛的颗粒及粉末所占百分比<15%符合标准, 3批次颗粒成型率为97.3%~98.5%, 符合规定; 休止角代表颗粒流动性, 一般认为休止角≤30°表明流动性良好, 3批次颗粒休止角在16.8°~18.0°, 表明颗粒流动性良好; 堆密度、 溶化性和水分均符合规定(表4).
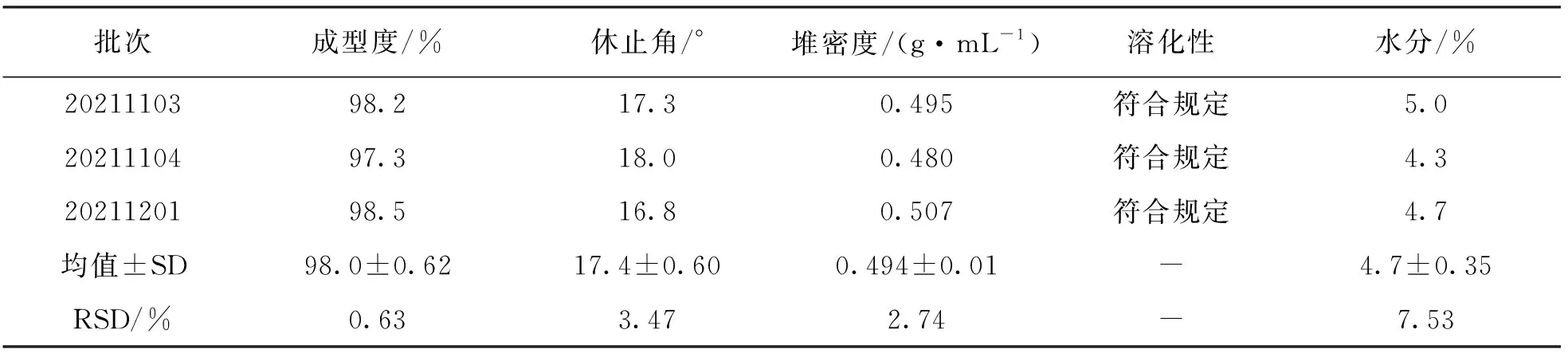
表4 六金颗粒3批次中试工艺验证生产结果
吸湿性检测7个时间点, 每24 h检测一次, 于24 h, 48 h, 72 h, 96 h, 120 h, 144 h和168 h分别测定颗粒的吸湿性, 并计算各时间点的吸湿率(表5). 测定结果表明, 3批次中试验证生产的六金颗粒吸湿拟合回归曲线方程为y=-0.000 92x+0.291 8x-1.219,r2=0.986 5, 拟合程度良好(图1), 平衡吸湿时间为162.1 h, 平衡吸湿率为24.6%.
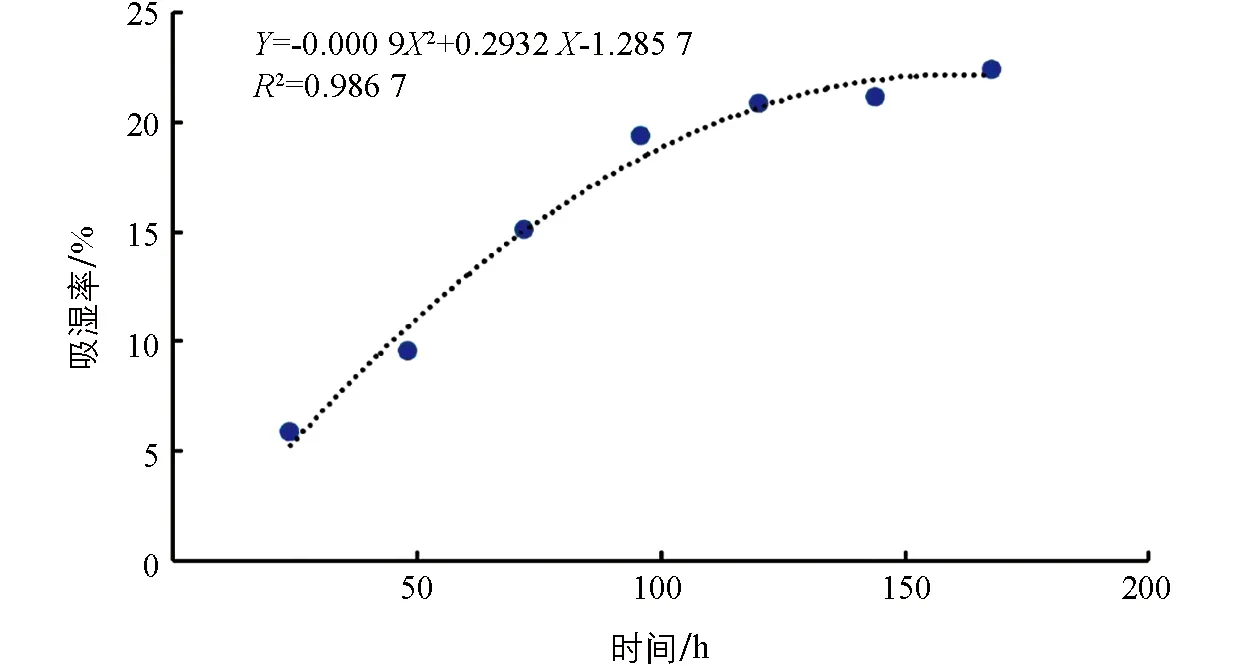
图1 六金颗粒吸湿曲线
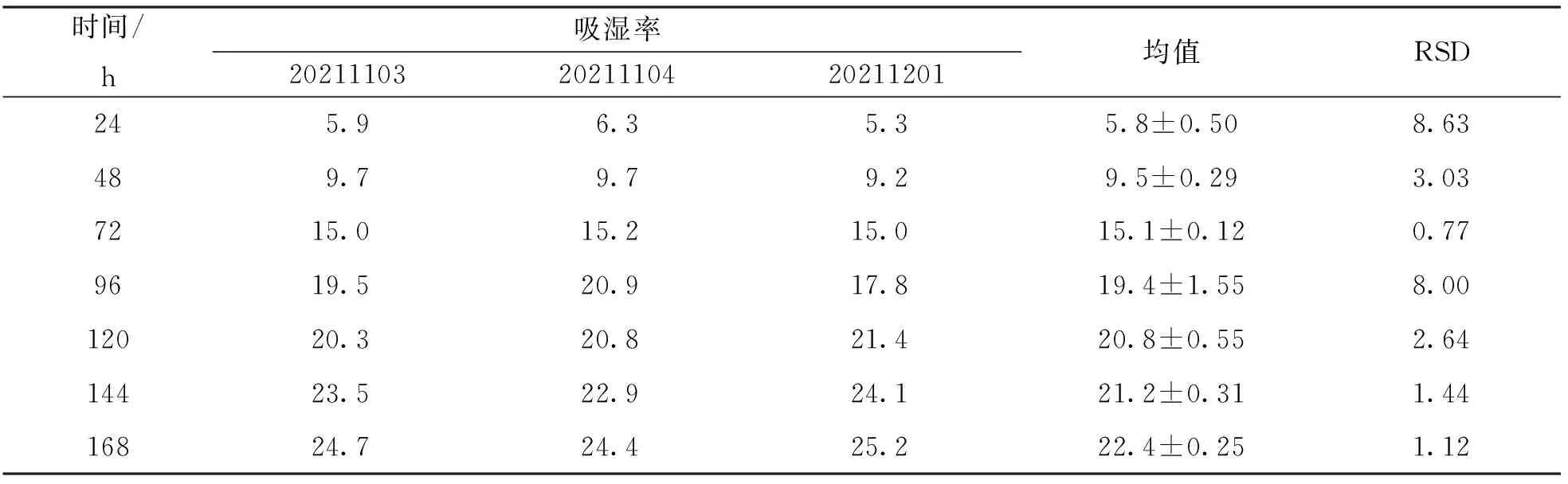
表5 六金颗粒3批次中试工艺验证吸湿率 %
3批次中试规模验证生产的六金颗粒符合《中国药典》2020年版四部通则0104颗粒剂项下检查要求.
2.2 锦灯笼薄层色谱鉴别
2.2.1 供试品溶液的制备
取本品3 g, 研细, 加甲醇25 mL, 超声处理30 min, 过滤, 滤液蒸干, 残渣加甲醇2 mL使其溶解, 作为供试品溶液.
2.2.2 对照品溶液的制备
取酸浆苦味素L对照品, 加甲醇制成每1 mL含0.1 mg的溶液, 作为对照品溶液.
2.2.3 锦灯笼阴性样品溶液的制备
取阴性样品3 g, 按2.2.1方法制备不含锦灯笼的六金颗粒阴性供试品溶液.
2.2.4 鉴别
分别吸取六金颗粒供试品溶液和锦灯笼阴性供试品溶液12 μL, 酸浆苦味素L对照品溶液10 μL, 在同一高效硅胶G薄层板上点样, 展开剂为三氯甲烷-丙酮-甲醇(25∶1∶1), 展开, 取出, 晾干, 显色剂为5%硫酸乙醇溶液, 在105 ℃加热至斑点清晰, 室温下放置30 min, 置紫外光灯365 nm下检视. 结果表明, 六金颗粒供试品色谱中, 在与对照品色谱相应的位置, 显相同黄色的荧光斑点, 阴性样品在相应位置无黄色荧光斑点, 说明该方法专属性良好(图2).
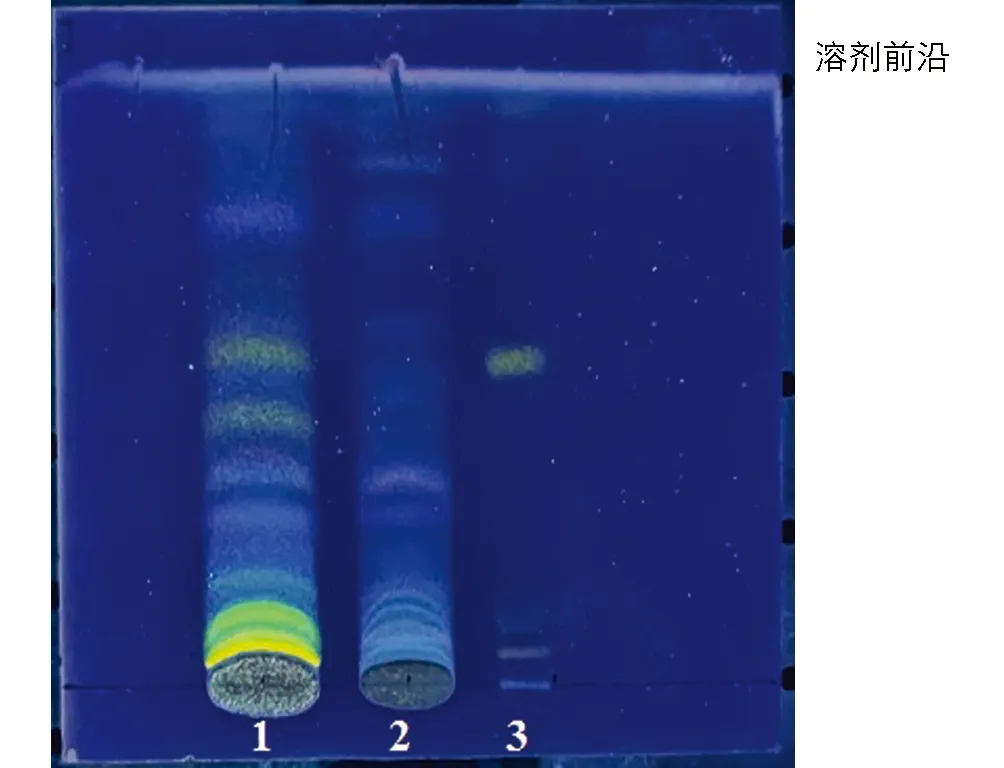
1: 供试品溶液; 2: 阴性样品溶液; 3: 酸浆苦味素L对照品溶液图2 锦灯笼薄层鉴别专属性试验色谱
2.2.5 样品检测
取不同批次六金颗粒(批号: 2202001, 2202002, 2202003), 按照“2.2.1”的方法制备供试品溶液, 按照“2.2.4”的方法进行鉴别. 结果表明, 3批六金颗粒供试品色谱中, 在与对照品色谱相应位置上, 显相同黄色的荧光斑点(图3).
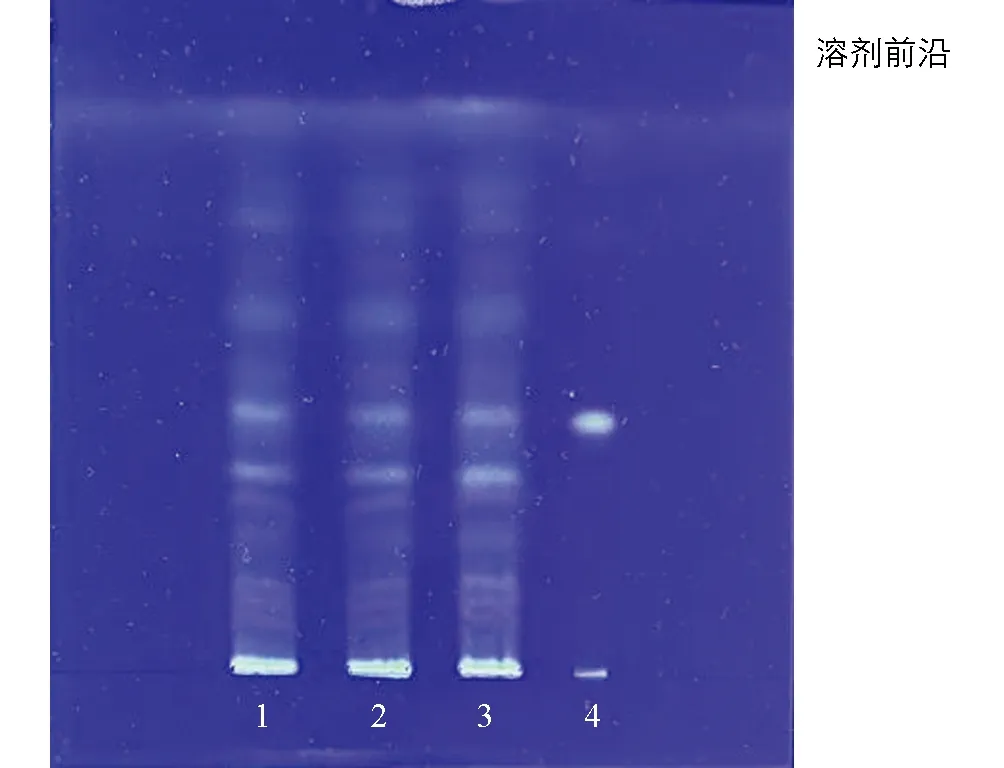
1: 供试品溶液(2202001); 2: 供试品溶液(2202002); 3: 供试品溶液(2202003); 4: 酸浆苦味素L对照品溶液图3 3批样品锦灯笼薄层鉴别色谱
2.3 蒲公英含量测定
2.3.1 色谱条件
以十八烷基硅烷键合硅胶为填充剂(4.6 mm×250 mm, 5 μm), 以乙腈-0.4%磷酸溶液(18∶82)为流动相, 检测波长为327 nm, 理论板数按菊苣酸峰计算应不低于5 000.
2.3.2 对照品溶液制备
取菊苣酸对照品适量, 精密称定, 加50%甲醇制成每1 mL含30 μg的溶液, 即得.
2.3.3 供试品溶液制备
取装量差异项下的本品, 混匀, 研细, 取约0.5 g, 精密称定, 置于具塞锥形瓶中, 精密加入50%甲醇25 mL, 称定重量, 超声处理(功率250 W, 频率40 kHz)30 min, 放冷, 再称定重量, 用50%甲醇补足减失的重量, 摇匀, 滤过, 即得.
2.3.4 蒲公英阴性供试品溶液的制备
取不含蒲公英的六金颗粒1.0 g, 精密称定, 按“2.3.3”项下方法制成蒲公英阴性供试品溶液, 即得.
2.4 蒲公英含量测定方法学考察
2.4.1 专属性
取菊苣酸对照品溶液、 六金颗粒供试品溶液、 蒲公英阴性供试品溶液各10 μL, 注入高效液相色谱仪, 按照“2.3.1”色谱条件进行样品检测. 结果表明, 在供试品色谱中, 在与对照品色谱相应的保留时间上有相同的色谱峰, 而蒲公英阴性供试品溶液无相应色谱峰; 表明处方中其他药材及辅料对菊苣酸含量测定结果无干扰, 表明该方法专属性良好(图4).
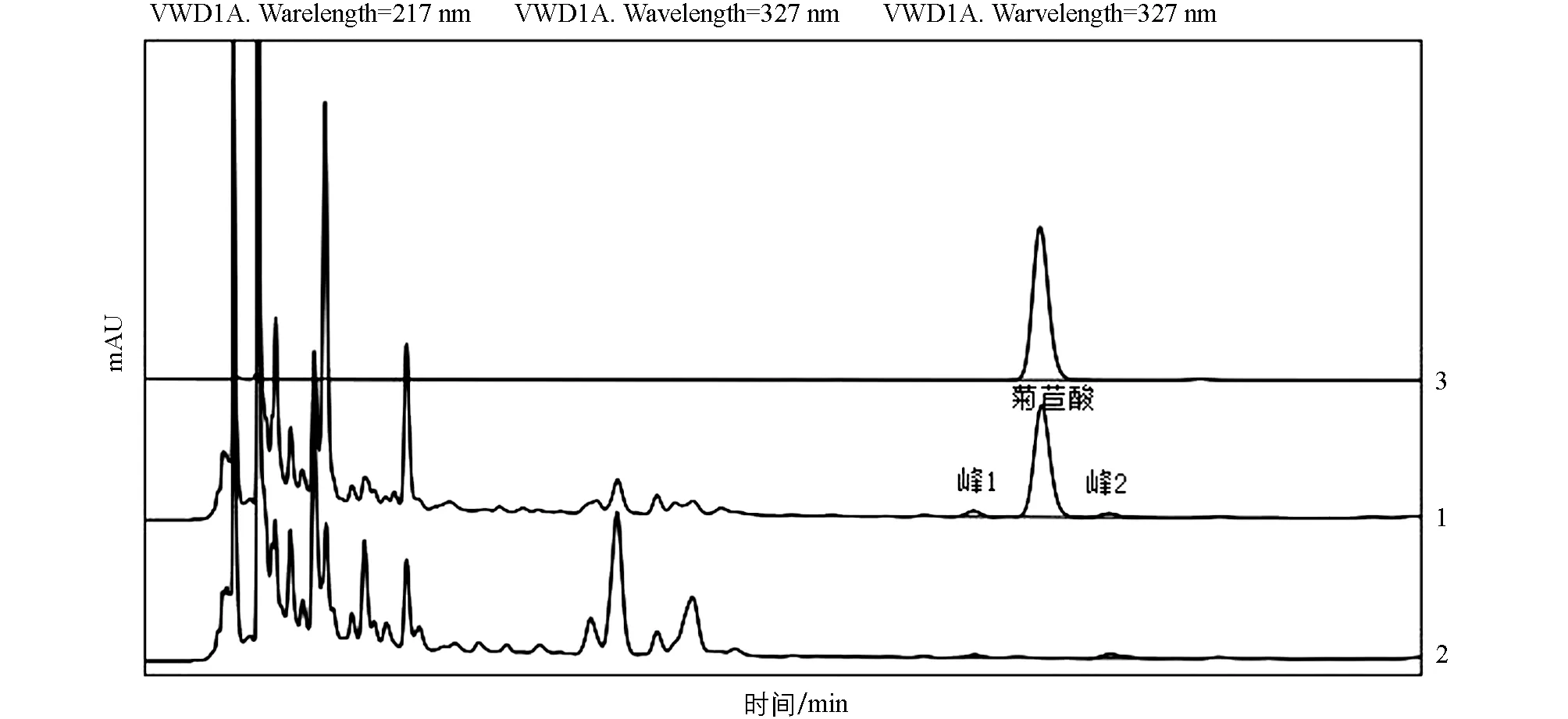
1: 六金颗粒供试品溶液; 2: 蒲公英阴性供试品溶液; 3:菊苣酸对照品溶液图4 菊苣酸含量测定方法学专属性色谱
2.4.2 线性试验
将浓度为718.28 μg/mL的菊苣酸对照品溶液, 分别稀释成浓度为1.44 μg/mL, 5.75 μg/mL, 14.37 μg/mL, 28.73 μg/mL, 57.46 μg/mL, 86.19 μg/mL的溶液, 按“2.3.1”项下色谱条件进样. 以菊苣酸的浓度为横坐标, 色谱峰面积为纵坐标, 进行线性回归, 得回归方程为y=45.196x-6.533 2, 相关系数为r=0.999 9, 菊苣酸在浓度1.44~86.19 μg/mL范围内, 线性关系良好.
2.4.3 重复性试验
取六金颗粒(批号: 2201001)平行制备6份供试品溶液, 按“2.3.3”项下方法制备供试品溶液, 按“2.3.1”项下色谱条件测定菊苣酸的质量分数, 得到菊苣酸的平均质量分数为1.28 mg/g, RSD为0.64%, 表明该方法重复性良好.
2.4.4 中间精密度试验
取六金颗粒(批号: 2201001), 由不同分析人员按“2.3.3”项下方法平行制备6份供试品溶液, 按“2.3.1”项下色谱条件采用不同仪器进行菊苣酸质量分数测定. 结果表明, 不同仪器的RSD为0.70%, 说明该方法所用仪器精密度良好.
2.4.5 稳定性试验
分别于第0 h, 12 h, 24 h, 48 h, 72 h进样测定同一六金颗粒供试品溶液(批号: 2201001), 计算菊苣酸的质量分数, 结果表明, 菊苣酸平均质量分数为1.30 mg/g, RSD为1.54%, 表明供试品溶液在72 h内稳定性良好, 均符合分析要求.
2.4.6 回收率试验
取六金颗粒6份, 分别加菊苣酸对照品溶液适量, 按“2.3.3”项下方法制备供试品溶液, 按“2.3.1”项下色谱条件测定菊苣酸的质量分数, 并计算加样回收率. 结果表明, 菊苣酸回收率为97.98%~101.32%(表6), RSD为1.36%, 符合分析要求.
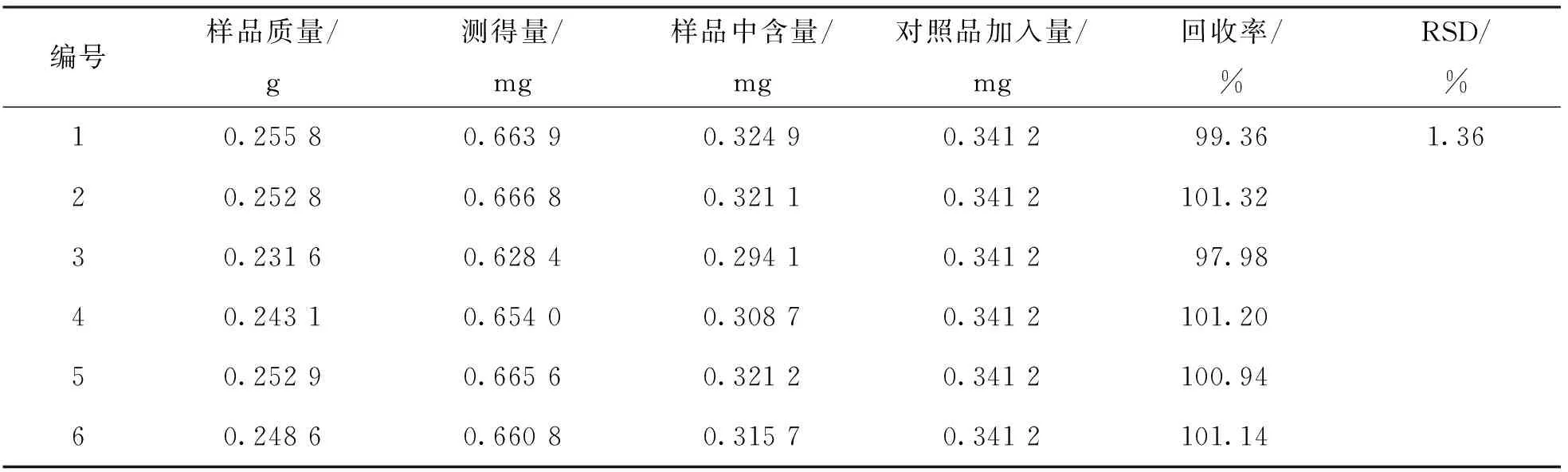
表6 回收率试验结果
2.5 HPLC特征图谱
2.5.1 色谱条件
以十八烷基硅烷键合硅胶为填充剂(Waters Atlantis T3, 4.6 mm×250 mm, 5 μm); 以甲醇为流动相A, 以0.2%磷酸为流动相B, 按表7的规定进行梯度洗脱; 检测波长为320 nm, 柱温为35 ℃. 理论板数按单咖啡酰酒石酸峰计算应不低于3 000.

表7 梯度洗脱表
2.5.2 对照品溶液制备
精密称取单咖啡酰酒石酸对照品, 加入50%甲醇, 制成每1 mL含30 μg的溶液, 即得.
2.5.3 供试品溶液制备
取装量差异项下的本品, 混匀, 研细, 取约0.5 g, 精密称定, 置具塞锥形瓶中, 精密加入10%甲醇25 mL, 称定重量, 超声处理(功率250 W, 频率40 kHz)30 min, 放冷, 再称定重量, 用10%甲醇补足减失的重量, 摇匀, 滤过, 取续滤液, 即得.
2.6 HPLC特征图谱方法学考察
2.6.1 重复性试验
取六金颗粒(批号: 2201001), 按“2.5.3”项下制备供试品溶液, 按“2.5.1”色谱条件测定, 结果表明, 8个特征峰的相对保留时间RSD≤0.61%, 相对峰面积RSD≤1.91%, 该方法的重复性良好.
2.6.2 稳定性试验
取六金颗粒(批号: 2201001), 按“2.5.3”项下制备供试品溶液, 按“2.5.1”色谱条件分别于0,3,6,12,24,48 h测定, 结果表明, 8个特征峰相对保留时间RSD≤0.45%, 相对峰面积RSD≤1.84%, 供试品溶液在48 h内稳定性良好.
2.6.3 中间精密度试验
取六金颗粒(批号: 2201001), 按“2.5.3”项下制备供试品溶液, 按“2.5.1”色谱条件测定, 结果表明, 8个特征峰的相对保留时间RSD≤0.85%, 相对峰面积RSD≤2.46%, 使用不同仪器的8个特征峰的相对保留时间RSD≤2.77%, 相对峰面积RSD≤2.57%, 该方法的中间精密度良好.
2.6.4 耐用性试验
取六金颗粒(批号: 2201001), 按“2.5.3”项下制备供试品溶液, 分别使用Waters Atlantis T3, Waters Xselect T3, Agilent TC-C18(2)不同色谱柱, 按“2.5.1”色谱条件测定, 结果表明, 8个特征峰的相对保留时间RSD≤3.87%, 相对峰面积RSD≤6.91%, 说明该方法适用性良好.
2.6.5 峰归属及峰指认
峰归属及峰指认结果表明, 峰1化合物未知, 属于龙脷叶药材; 峰2化合物未知, 属于忍冬藤药材; 峰3化合物为新绿原酸, 属于蒲公英药材; 峰4化合物为单咖啡酰酒石酸, 属于蒲公英药材; 峰5化合物未知, 属于忍冬藤药材和龙脷叶药材; 峰6化合物为绿原酸, 属于忍冬藤药材、 蒲公英药材、 龙脷叶药材; 峰7化合物未知, 属于忍冬藤药材、 蒲公英药材、 锦灯笼药材; 峰8化合物为隐绿原酸, 属于忍冬藤药材(图5).
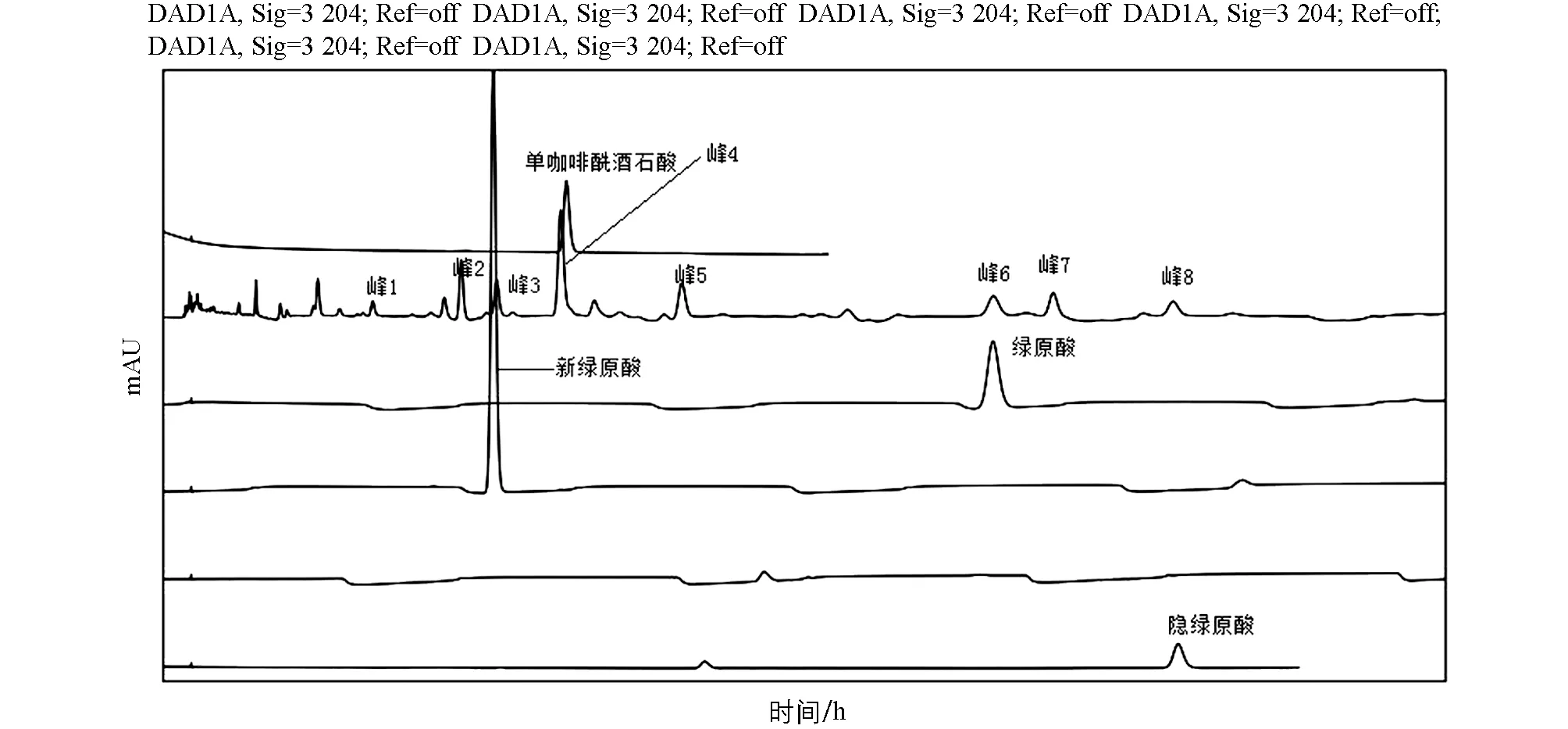
图5 六金颗粒特征图谱峰指认结果
2.7 样品测定
取不同批次六金颗粒(批号: 2202001,2202002,2202003), 按“2.3.3”项下方法制备供试品溶液, 按“2.3.1”色谱条件测定菊苣酸的质量分数, 结果显示3批样品中(每袋装10 g)菊苣酸平均质量分数为1.32 mg/g, RSD为0.44%. 按“2.5.3”项下方法制备供试品溶液, 按“2.5.1”色谱条件测定, HPLC特征图谱8个特征峰的相对保留时间RSD≤1.02%, 相对峰面积RSD≤3.55%, 结果符合规定.
3 结论与讨论
中药颗粒剂是在中药汤剂和糖浆等剂型的基础上发展起来的新剂型, 既保持了汤剂吸收快、 作用迅速的特点, 又克服了汤剂使用时煎煮不便、 服用量大、 易霉变质等缺点, 较之注射剂等能较大程度地保留药材有效成分, 制备工艺相对简单, 能适用于工业生产, 剂量小, 服用、 携带、 储藏、 运输均较方便[9-10], 故在剂型选择时确定为颗粒剂.
六金颗粒在喷干粉储存过程中发现物料容易吸湿结块, 加入辅料可以明显改善吸湿结块问题[11]. 本研究选择中药制粒工艺中常用的辅料可溶性淀粉、 淀粉、 麦芽糊精、 糊精和乳糖[12]进行了比较研究. 因本品喷干粉本身吸湿性较强, 因此加入乳糖后增加了其吸湿性, 黏筛网无法进行制粒. 可溶性淀粉和淀粉具有良好的抗湿性, 但是在制粒过程产生的细粉过多, 使得颗粒成型率低. 麦芽糊精和糊精具有良好的成型性, 但糊精的溶化性很差, 形成白色沉淀, 最终选择使用麦芽糊精降低了喷干粉的水分含量并改善了其吸湿性.
六金颗粒处方中含有锦灯笼, 锦灯笼中主要化学成分为酸浆苦味素类, 使得颗粒口感偏苦, 影响了患者的顺应性. 对三氯蔗糖、 甜菊糖苷和阿斯巴坦3种矫味剂进行了筛选, 除了口感上的比较之外, 还考虑了安全性因素. 阿司帕坦不适用于苯丙酮酸尿患者[13]; 三氯蔗糖会影响肠道微生物群的平衡, 促使炎症因子的富集[14]; 甜菊糖苷未见不良反应报道, 加之可以有效改善口感, 因此本品选择甜菊糖苷作为矫味剂.
制剂的活性成分能反应制剂的有效性, 对工艺路线选择、 成型工艺、 制剂稳定性有监控作用, 故处方制剂中已明确有活性成分的首先选择活性成分进行质量标准研究, 同时选择指标时还应重视对制剂质量的可控性, 对于含量低于制剂万分之一的成分已基本无质控意义[15-16]. 六金颗粒处方有效成分复杂, 蒲公英作为君药, 具有广泛的生物活性, 起到清热解毒功效. 根据“君臣佐使”与药效活性相结合的原则, 本研究中含量测定指标化合物定为菊苣酸, 采用高效液相色谱法对制剂中的菊苣酸进行含量测定, 对流动相比例反复优化, 最终确定了最优的流动相比例及洗脱梯度, 结果表明该方法易于操作, 重复性良好.
中药指纹图谱技术是从中药整体化学物质基础角度出发, 对中药化学成分特征进行系统的、 整体的、 专属的表征的方法, 具有专属性强、 稳定性好、 重现性好的特点[17-18]. 六金颗粒采用HPLC法建立了特征图谱, 通过特征峰的相对保留时间及色谱峰的数量来检测不同批次间制剂的差异, 可作为制订前端药材内控标准的依据.
鉴于中药成分复杂、 基础研究薄弱、 生产过程复杂等特点, 质量标准研究难以从全成分检测和控制上设置相应检测项目. 《中药新药质量标准研究技术指导原则(试行)》[19]提出根据药品的特点建立专属性、 针对性的检查方法建立反映制剂特点的检查项目, 鼓励指纹图谱或特征图谱应用于中药这种复杂体系的含量测定, 从而提高质量标准的整体可控性. 本研究目前仍采用较为传统的药品质量控制模式[20], 对蒲公英、 忍冬藤等6味药汤剂转化为颗粒剂, 进行了制备工艺研究和质量标准研究, 所建立的六金颗粒制备工艺稳定合理可行, 建立的质量标准中薄层色谱鉴别、 含量测定和特征图谱方法科学、 合理, 能够多角度地有效控制产品质量, 为后续产品生产及品质评价提供参考依据. 随着多学科互动协作, 新技术、 新方法, 如质-量双标法、 谱效关系法、 基于网络药理学及代谢组学的质量标志物法等多种模式和手段的应用[18, 21], 将为中药制剂质量控制研究带来新的发展思路.
