N-gamboyl Gemcitabine Inhibits Tumor Cells Proliferation and Migration
2023-09-22PEIYifei裴屹斐DENGMinJIANGYuxin姜玉新SHAOZhiyu邵志宇WANGHongsheng王红声
PEI Yifei(裴屹斐), DENG Min(邓 敏), JIANG Yuxin(姜玉新), SHAO Zhiyu(邵志宇), WANG Hongsheng(王红声)
1 Shanghai Engineering Research Center of Nano-Biomaterials and Regenerative Medicine, College of Biological Science and Medical Engineering, Donghua University, Shanghai 201620, China 2 College of Chemistry and Chemical Engineering, Donghua University, Shanghai 201620, China 3 The First Hospital of Jiaxing, Jiaxing Key Laboratory of Virus-Related Infectious Diseases, Jiaxing University, Jiaxing 314001, China
Abstract:Gambogic acid (GA) is a natural substance with a good antitumor effect, but it is too lipophilic to be metabolized and excreted, thus accumulating in the body. Gemcitabine (GEM), one of the first-line antitumor drugs, has high hydrophilicity, which greatly shortens its half-life in vivo. We previously reported a compound named N-gamboyl gemcitabine (GAG), derived from the condensation of GEM and GA, whose hydrophilicity is better than GA and stability is better than GEM. Here, the antitumor performance of GAG was investigated for the first time by using several common tumor cell lines as tumor models. The results of in vitro study showed that GAG significantly inhibited the proliferation and migration of the tumor cells. The IC50 values of GAG for the tumor cells were lower than those of GEM and GA. The present study suggests that GAG has a promising potential to be developed into a broad-spectrum antitumor drug.
Key words:N-gamboyl gemcitabine (GAG); gambogic acid(GA); gemcitabine (GEM); antitumor
0 Introduction
Cancer seriously endangers human health and imposes a huge social burden on human beings. According to the latest statistics released by the National Cancer Center of China, the number of cancer deaths in the world is as high as 9.96 million per year, of which China accounts for 30% of the total number of cancer deaths with 3 million, becoming the country with the largest number of cancer deaths in the world[1-2].
Natural compounds show a wide range of potential in the treatment of various diseases. They have high biological activity, can effectively inhibit pathogens, and have good biocompatibility with fewer side effects[3]. Gambogic acid (GA), a dry resin derived from the Garcinia cambogia plant, has good antitumor activity[4]. However, GA is very lipophilic, which makes it easy to accumulateinvivoand thus not suitable for use in clinic[5-6]. Gemcitabine (GEM) has good hydrophilicity and is currently one of the first-line drugs for the clinical treatment of malignant tumors such as pancreatic cancer and cervical cancer[7-8]. However, the structural defect of GEM makes it easy to be deaminated by cytosine deaminase in the human body, resulting in a short half-life and the patients have to take a large dose and thus bear a greater burden[9]. Too high lipophilicity or hydrophilicity could affect the efficacy of the drug. Therefore, maintaining the appropriate amphiphilicity can improve the pharmacokinetic properties of the drug in the human body, increase the druggability of the compound, and achieve the desired therapeutic effect. It is a good idea to develop an antitumor drug with good biocompatibility and amphiphilicity based on the advantages of both GEM and GA. By condensation of GEM and GA, we developed an amide derivative of GA, namedN-gamboyl gemcitabine (GAG, CAS:1453868-27-3) which may meet the requirement of ideal drugs[10]. In this study, we investigated the antitumor activity of GAG for the first time by using common malignant tumor cells including pancreatic cancer cells (PANC-1), liver cancer cells (HepG-2), melanoma cells (B16) and cervical cancer cells (HeLa) as tumor models. The present work lays a foundation for evaluating the clinical application potential of GAG.
1 Experiments
1.1 Materials
GEM was purchased from Beijing Yinuokai Technology Co., Ltd., Beijing, China. Dulbecco’s modified Eagle’s medium (DMEM) was purchased from Gicob Company, USA. GA is synthesized by our group. The 1640 medium was purchased from Shanghai Yuanye Biotechnology Co., Ltd., Shanghai, China, and the trypsin digestion solution with a mass fraction of 0.25% was purchased from Gibco, USA. Phosphate buffered solution (PBS, pH=7.2) was purchased from Wuhan Sewell Biotechnology Co., Ltd., Wuhan, China. Dimethyl sulfoxide (DMSO),N,N-dimethylformamide (DMF), ethyl acetate, sodium chloride, anhydrous sodium sulfate and deuterated chloroform (CDCl3) were purchased from China National Pharmaceutical Group Co., Ltd., Beijing, China. Fetal bovine serum was purchased from Zhejiang Tianhang Biotechnology Co., Ltd., Hangzhou, China. CCK-8 kit was purchased from Sangon Bioengineering Co., Ltd., Shanghai, China. 1-hydroxybenzotriazole (HOBT) and 1-ethyl-3-(3-dimethylaminopropyl) carbodiene imine hydrochloride (EDCI) were purchased from Sigma, USA.
1.2 Preparation and characterization of GAG
GAG was prepared according to our patent[10]. Briefly, GA, GEM, HOBT, and EDCI are dissolved in DMF at room temperature, stirred and reacted at (50±10) ℃ for 3 h. After the reaction was completely completed, ethyl acetate was added to quench the reaction, and then the resulting mixture was washed several times with the saturated sodium chloride solution. After the resulting mixture was dried with anhydrous sodium sulfate, ethyl acetate was removed by evaporation. Finally, the crude product was eluted by column chromatography to obtain GAG. The product (10 mg) was dissolved in CDCl3reagent (0.6 mL) and placed in a nuclear magnetic tube for nuclear magnetic resonance (NMR) testing with an NMR instrument (AV-400, Brucker, Switzerland). Tetramethylsilane was used as an internal reference and the chemical shift multiplied by 106was represented byδin this paper.
1.3 Cell proliferation and toxicity experiments
B16 cells were cultured in 1640 medium with 10% (mass fraction) fetal bovine serum and 1% (mass fraction) penicillin/streptomycin. PANC-1, HepG-2, HeLa, and L929 cells were cultured in DMEM with 10% (mass fraction) fetal bovine serum and 1% (mass fraction) penicillin/streptomycin. The above cells were seeded into 24-well tissue culture plates and cultured in a constant temperature incubator at 37℃, and the volume fraction of carbon dioxide was controlled at 5%. A certain amount of DMSO was used to preliminarily dissolve the drug (GAG, GEM, or GA) to prepare the stock solution. The stock solution was then diluted with PBS and added to the medium culturing PANC-1, HepG-2, B16, and HeLa, respectively. The final concentration of the drug in the cell-cultured medium was 50, 10, 1, 0.1 and 0.01 μmol/L. The final DMSO mass fraction in the cell-cultured medium did not exceed 0.2%. After the treatment with the drug for 24 h, the cells were washed three times with PBS, and 200 μL of 1640 medium (for B16) or DMEM (for PANC-1, HepG-2, and HeLa) with 10% (mass fraction) CCK-8 reagent was added to each well and continued to incubate in the dark for 1 h. Then, 100 μL of supernatant was transferred to a 96-well plate, and the absorbance value was measured at a wavelength of 450 nm by using a microplate reader (Thermo Fisher Company, USA). Finally, the cell viabilityVcellwas calculated according to
(1)
whereAtis the absorbance of the tested sample;Abis the absorbance of the blank sample containing culture medium and CCK-8 solution but without cells;Acis the absorbance of the control sample.
Also, L929 cells were used as a model of normal cells to investigate whether the drugs (at a certain concentration based on the above results) affect the proliferation of non-tumor cells.
1.4 Assays for detection of cell apoptosis
1.4.1Hoechst/propidineiodide(PI)doublestainingassay
PANC-1, HepG-2, B16, HeLa and L929 cells were seeded in a 48-well tissue culture plate and cultured as described above. GAG was used to treat the cells at a final concentration of 1 μmol/L for 24 h. Then, 200 μL of Hoechst 33258 staining solution (5 μg/mL) was added to stain the cells in the dark for 10 min. Subsequently, 200 μL of PI staining solution (15 μg/mL) was added to react at 4 ℃ for 10 min in the dark. Finally, cells were observed and taken pictures with an inverted fluorescent microscope.
1.4.2Westernblotassay
PANC-1, HepG-2, B16 and HeLa cells were cultured in a six-well tissue culture plate. When the cell confluence was up to about 90%, the GAG was used to treat the cells at a final concentration of 1 μmol/L, and the cells treated with the same amount of PBS were set as the control. After incubation for 48 h, the cells were then harvested and the total protein was extracted from them by using RIPA lysate. The protein concentration was measured, and the samples were denatured in a water bath for 15 min before being subjected to SDS-PAGE gel electrophoresis. After being transferred to a polyvinylidene fluoride membrane, the sample was treated overnight with the first antibodies (anti-Bax and anti-Caspase 9) at 4 ℃, and then incubated at room temperature for 30 min with the second antibody. Subsequently, the enhanced chemiluminescence solution was added to fully react for 2 min, and then the membrane was imaged with a chemiluminescence instrument (ChemiScope 6100BZ, Clinx, Shanghai, China).
1.5 Cell scratch assay
PANC-1, HepG-2, B16 and HeLa cells were seeded respectively in a 24-well tissue culture plate with 150 000 cells per well. After the cells were cultured for 24 h, a 200 μL pipette tip was used to draw a trace in the middle of the bottom of each well. Then GAG was added to the wells at a concentration of 1 μmol/L (PBS was used as the control) and the cells were incubated for another 24 h. Then, the cells were photographed and the cell migration rate was calculated.
1.6 Statistical analysis
All experiments were conducted at least three times, and numeric data were expressed as mean ± standard deviation (SD) unless otherwise indicated. Statistical significance was analyzed by one-way analysis of variance (ANOVA) with Tukey’s post hoc test or unpairedt-test. Ap-value less than 0.05 was considered statistically significant.
2 Results and Discussion
2.1 1H NMR of GAG
The product obtained by the condensation reaction of GA and GEM was a yellow compound named GAG. The molecular structure of GAG had groups from both GA and GEM (Fig. 1), which was proved by the1H NMR spectrum (Fig. 2). In Figs. 1 and 2, red letters were used in labeling H. The1H NMR data of GAG are as follows.
1HNMR(400 MHz, CDCl3)δ:12.80 (s, 1 H, Ha), 8.20 (s, 1 H, Hb), 8.13 (d,J= 6.7 Hz, 1 H, Hc), 7.55 (d,J= 6.9 Hz, 1 H, Hd), 7.33 (d,J= 7.6 Hz, 1 H, He), 6.55 (d,J= 10.2 Hz, 1 H, Hf), 6.20 (t,J= 6.0 Hz, 1 H, Hg), 5.80 (t,J=8.4 Hz, 1 H, Hh), 5.40 (d,J= 10.2 Hz, 1 H, Hi), 5.05-4.94 (m, 2 H, Hjand Hk), 4.62-4.43 (m, 1 H, Hl), 3.57-3.44 (m, 2 H, Hm).
As shown in Fig. 2, the characteristic peaks of the1H NMR spectrum were assigned according to the structure, in which the ratio of the GA part and the GEM part was about 1∶1, and the peaks of the protons on the hydroxyl carbon were atδof 4.62-4.43 and 3.57-3.44 without significant change. Therefore, it is determined that the condensation reaction on the hydroxyl group did not occur. The single peak atδof 8.20 was assigned to a new amide hydrogen (Hb), indicating that GA and GEM were condensed through the amide bond to obtain GAG. The peaks appearing atδof 1-2 were originated from 8 methyl groups of GA.
Drugs with amphiphilic properties, that is, having both hydrophilicity and lipophilicity, are more beneficial to human absorption[11-12]. GA is a compound with too strong lipophilic, which greatly limits its clinical application[13]. GEM is unstable and easy to be degradedinvivodue to its high hydrophilicity, which gives it a very short half-life[14-17]. GAG has the characteristics of both GA and GEM in structure, that is, it has both hydrophilicity and lipophilicity to a certain extent. Hence, GAG is more in line with the requirements of ideal drugs since it avoids the structural defects of GA and GEM.
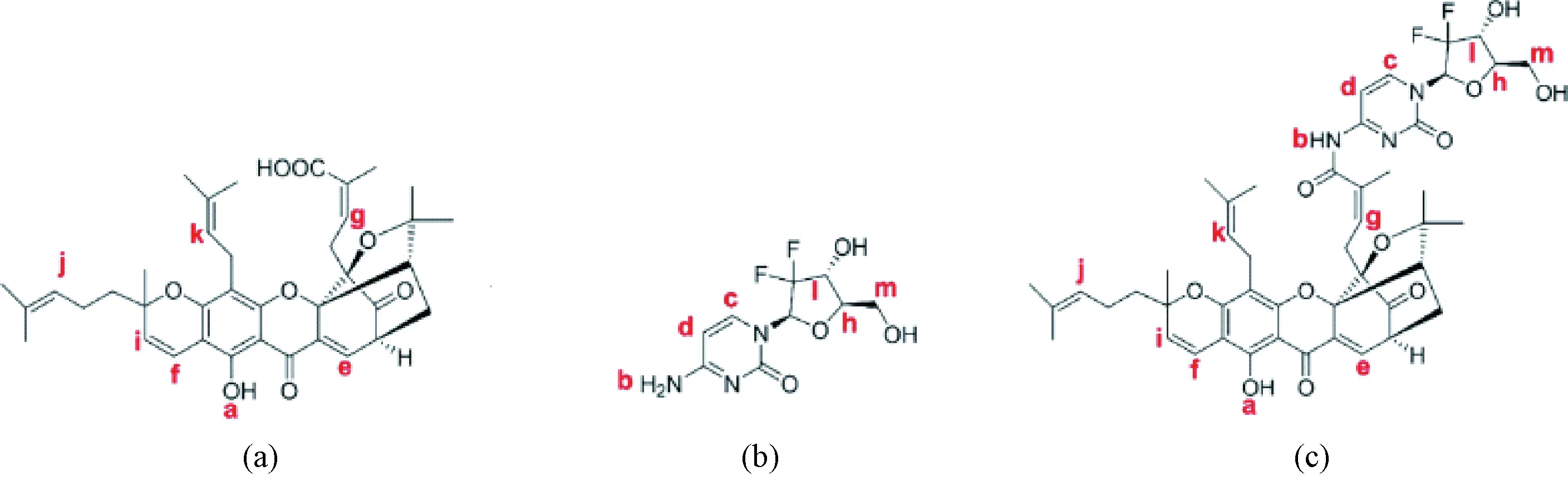
Fig. 1 Molecule structures of the compounds:(a) GA; (b) GEM; (c) GAG
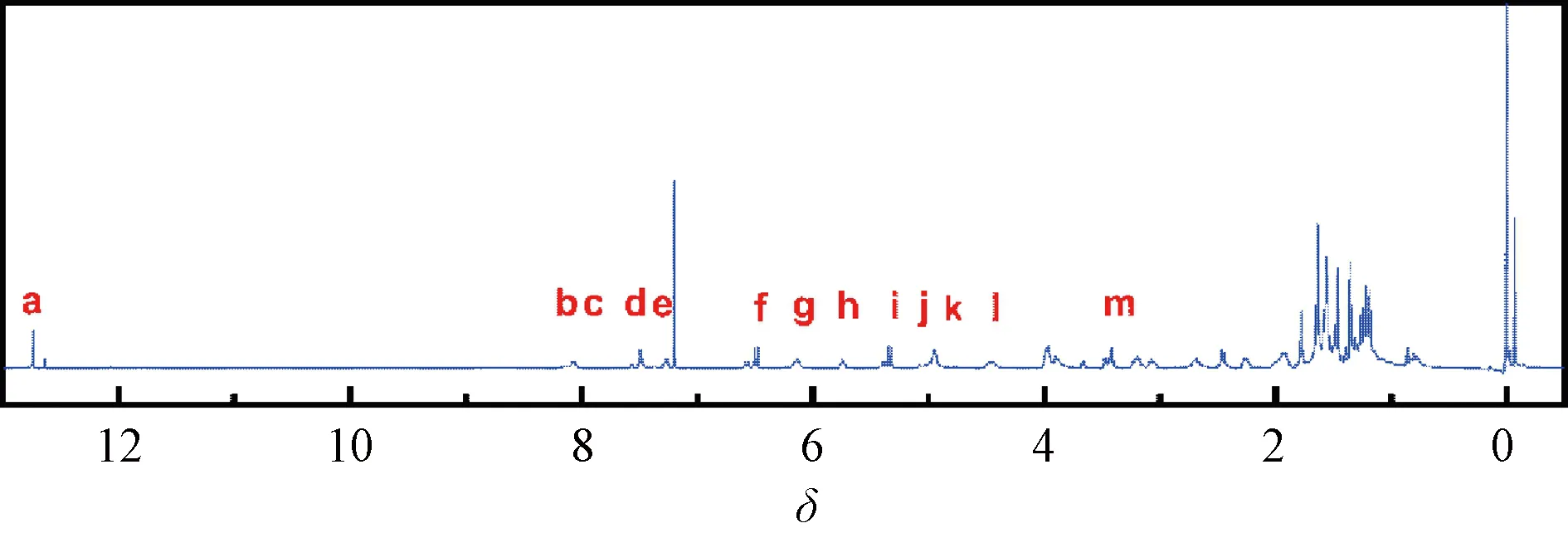
Fig. 2 1H NMR spectrum of GAG
2.2 Effect of GAG on the proliferation of tumor cells
To investigate whether GAG inherits the antitumor activity of GA and GEM, the cytotoxicity of GAG on tumor cells was detected by using several cell lines of common tumors (PANC-1, HepG-2, B16 and HeLa) as models. As shown in Fig. 3, all three drugs can significantly inhibit the proliferation of the selected tumor cells in a concentration-dependent manner. Moreover, the IC50 value of GAG for the tumor cells is less than 1 μmol/L, lower than that of GA and GEM (Table 1), indicating that GAG has the best performance in inhibiting tumor cells among the three drugs. What is interesting is the antitumor mechanism of GAG, since it has such a powerful effect. GA has been reported to enhance the expression of tumor necrosis factors and inhibit the expression of genes involved in apoptosis, thereby inducing the apoptosis of tumor cells[18]. GEM blocks the proliferation of tumor cells by embedding in the DNA chain to terminate the synthesis of DNA[16]. It is speculated that the antitumor mechanism of GAG is similar to that of GA and GEM, which needs to be confirmed by further research.
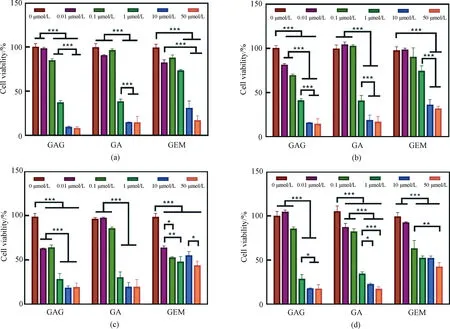
*—— p <0.05; **—— p <0.01; ***—— p<0.001Fig. 3 Effects of GAG, GA and GEM on several cell lines of tumor cells proliferation:(a) PANC-1; (b) HepG-2; (c) B16; (d) HeLa

Table 1 IC50 values of the drugs for killing tumor cells Unit:μmol/L
Based on the above results, we further analyzed the effects of different drugs on the growth of different cells (including tumor cells and normal cells) at the same concentration of 1 μmol/L. As shown in Fig. 4, GAG and GA had similar killing effects on tumor cells and were better than GEM. Furthermore, all the three drugs showed no significant adverse effects on the cell viability of L929, indicating that the drugs had good biosafety within their effective antitumor concentration range.
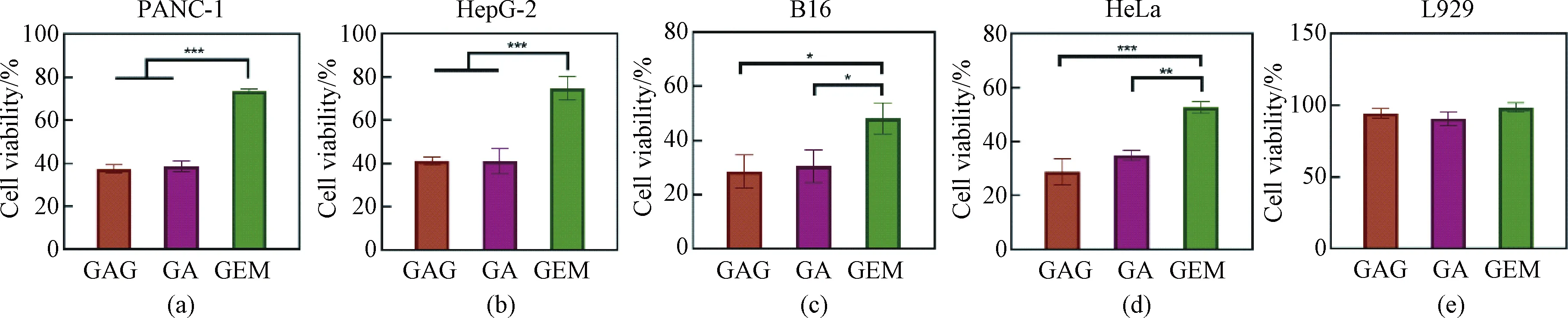
Fig. 4 Effects of GAG, GA and GEM at the concentration of 1 μmol/L on the proliferation of different cells:(a) PANC-1; (b) HepG-2; (c) B16; (d) HeLa; (e) L929
2.3 Effect of GAG on apoptosis
In order to explore the mechanism of GAG’s inhibition of tumor cell growth, we investigated whether this compound had the ability to induce tumor cell apoptosis. The Hoechst/PI double staining method was used for the detection of apoptosis of the cells treated with GAG at the concentration of 1 μmol/L. Normal cells and early &mid-stage apoptotic cells can be stained by Hoechst 33258, with normal cells stained lighter color and the apoptotic cells brighter color[19]. The cells that can be stained by PI are necrotic cells or advanced apoptotic cells[19]. As shown in Fig. 5, the cells in the control group were stained light blue by Hoechst 33258, and few cells were stained red by PI staining, which indicated that these cells grew well, with rare apoptosis. While for the tumor cells treated with 1 μmol/L GAG, a large number of cells were stained bright blue by Hoechest 33258 or red by PI, indicating massive apoptosis. However, for the normal cells (L929) treated with GAG, there was almost no evidence of apoptosis, and there was no significant difference compared with the control group. These data demonstrate that 1 μmol/L of GAG can effectively inhibit tumor cell growth by inducing their apoptosis, and at this concentration, GAG has no significant toxicity to normal cells.
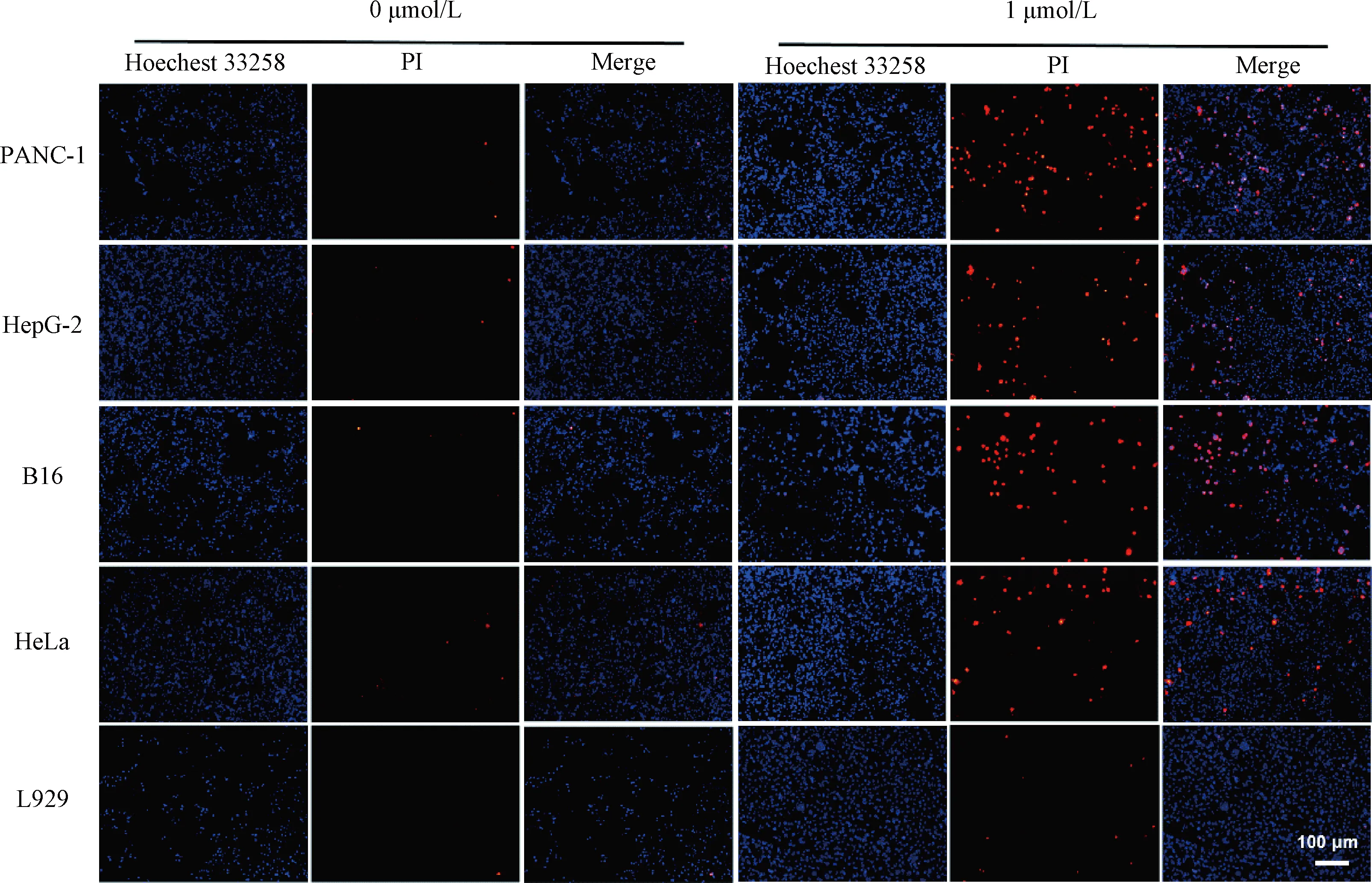
Fig. 5 Hoechst/PI double staining of the cells treated by GAG at a concentration of 1 μmol/L
Apoptosis is a complex physiological process involving the regulation of many related genes including the Bcl-2 family and the caspase family. Bax, a key member of the Bcl-2 family, is a pro-apoptotic protein, the increase of its level would enhance the apoptosis of cells[20-21]. Caspase 9, a protein that can induce apoptosis by cleaving substrates such as poly (ADP-ribose) polymerase after activation, also plays a key role in the entire process of apoptosis[22-23]. To explore the mechanism of GAG-induced apoptosis, the levels of Bax and Caspase 9 were detected in the tumor cells after treatment with 1 μmol/L of GAG by western blot. Glyceraldehyde-3-phosphate dehydrogenase(GAPDH) was used as an internal reference for western blot protein normalization. As shown in Fig. 6, the expression levels of both Bax and Caspase 9 were significantly increased in the tumor cells treated with GAG, compared with the control, which suggested that GAG-induced apoptosis of the tumor cells was related to promoting the expression levels of these two proteins. GA has also been reported to induce apoptosis of various cancer cells by increasing the expression of Bax and Caspase 9[24]. Furthermore, GA can also directly target mitochondria, causing rapid depolarization and fragmentation of mitochondrial membranes, and causing the release of cytochrome C[25]. The mechanism of GEM-induced tumor cell apoptosis mainly lies in its incorporation into the DNA chain[26]. Therefore, the mechanism of GAG-induced tumor cell apoptosis could also be related to mitochondria-dependent apoptosis and destruction of chromosome structure since GAG shares the structure of both GA and GEM, which needs to be verified by further study.
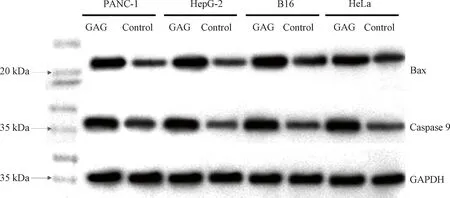
Fig. 6 Expression of Bax and Caspase 9 in tumor cells treated with GAG
2.4 Effect of GAG on cells scratch healing
GA has been reported to significantly inhibit the invasion and metastasis of tumor cells by suppressing TGF-β1-induced epithelial-mesenchymal transition[27]. While GEM can inhibit tumor cell migration and invasion by inducing the expression of E-cadherin and reducing the levels ofN-cadherin, Snail, vimentin and proangiogenic factors (including VEGF, VEGFR2, CD31 and HIF-1α)[28]. To explore whether GAG can also inhibit the migration of tumor cells, aninvitroscratch healing experiment was conducted, wherein the scratch healing area of the tumor cells was observed and calculated after treatment with 1 μmol/L of GAG for 24 h. As shown in Fig. 7, the scratch healing areas in the cells treated with GAG were significantly smaller than that in the control, which suggested that GAG effectively inhibited the migration of the tumor cells. Taken together, our data suggest that GAG has the potential to be developed into a better antitumor drug than GA and GEM.

Fig. 7 Effect of GAG at a concentration of 1 μmol/L on the migration of tumor cells:(a) PANC-1; (b) HepG-2; (c) B16; (d) HeLa
3 Conclusions
In the present study, the excellent antitumor activity of GAG derived from the condensation of GEM and GA was confirmed for the first time. GAG was demonstrated to inhibit the growth of various tumor cells, with lower IC50 values than those of GEM and GA. Our data also show that the antitumor mechanism of GAG is related to promoting the expression of pro-apoptotic proteins. Considering that its antitumor performance and stability are better than GEM, GAG has great application potential in clinic use as a broad-spectrum antitumor drug candidate.
杂志排行

Journal of Donghua University(English Edition)的其它文章
- Image Retrieval with Text Manipulation by Local Feature Modification
- Multi-style Chord Music Generation Based on Artificial Neural Network
- Polypyrrole-Coated Zein/Epoxy Ultrafine Fiber Mats for Electromagnetic Interference Shielding
- Design of Rehabilitation Training Device for Finger-Tapping Movement Based on Trajectory Extraction Experiment
- High-Efficiency Rectifier for Wireless Energy Harvesting Based on Double Branch Structure
- Review on Development of Pressure Injury Prevention Fabric