电石渣对碱激发粉煤灰-矿渣抗碳化性能的影响
2023-09-22肖欣欣王柏文鲁乃唯
刘 扬,肖欣欣,2,陈 湘,2,王柏文,2,罗 冬,2,鲁乃唯
(1.长沙理工大学土木工程学院,长沙 410114;2.长沙理工大学桥梁工程安全控制教育部重点实验室,长沙 410114)
0 引 言
硅酸盐水泥在生产过程中存在明显的资源消耗和环境污染问题,碱激发凝胶材料作为一种新型的绿色环保建筑材料[1],可替代硅酸盐水泥实现工业可持续发展。然而,碱激发凝胶材料的应用受到较大限制,主要原因之一是目前其耐久性能的研究不够完善[2],抗碳化性能是衡量材料耐久性能的一个重要指标[3]。
当空气中的CO2从材料表面入侵至内部时,会使材料发生碳化反应,碱度由外到内降低,加剧钢筋锈蚀的风险。黄琪等[4]研究发现,经碳化作用后普通混凝土和低钙粉煤灰基地聚物的微观结构变得更加致密,但碱激发粉煤灰-矿渣碳化后的孔隙变化则完全相反[5]。在碳化过程中,材料的孔隙结构变化影响着碳化深度的发展,Huang等[6]通过研究发现,不同碱激发复合凝胶材料的碳化深度远大于普通硅酸盐水泥的碳化深度。
碳化对碱激发凝胶材料的影响主要是导致其碱度与强度的变化。目前测试碱度时常用的方法是浸泡试件粉末后测量 pH 值[7-8]。在加速碳化的条件下,碱激发凝胶材料的碱度低于硅酸盐水泥的碱度,前者碱度下降速率更大[5-6]。采用低钙粉煤灰制备的粉煤灰基地聚物初始碱度较低,掺入矿渣后可提高其初始碱度[9];与低钙碱激发凝胶材料相比,高钙碱激发凝胶材料被钢筋锈蚀的风险更大[10]。除此之外,碱激发粉煤灰-矿渣经碳化作用后,会发生水化硅(铝)酸钙(C-(A)-S-H)凝胶脱钙的现象[11],导致抗压强度降低[5],而碱激发凝胶材料经碳化后强度下降被归因于孔结构粗化[12],这意味着高钙碱激发粉煤灰-矿渣虽然具备更高的抗压强度,但是抗碳化耐久性能存在较多不足。
碱性物质的供应是预防材料碳化侵蚀的首要措施[13]。电石渣是电石水解过程中产生的强碱性废渣,主要成分为Ca(OH)2[14]。在碱激发粉煤灰-矿渣的原料中掺入电石渣,不仅可以为体系提供碱性储备,还能进一步提升碱激发粉煤灰-矿渣的抗压强度[15]。因此,本文以电石渣部分替代粉煤灰掺入碱激发粉煤灰-矿渣(alkali-excited fly ash-slag, AAFS)来制备粉煤灰-矿渣-电石渣复合凝胶材料(alkali-excited fly ash-slag composite gel material, AAFSC),探究电石渣的掺入对其抗碳化性能的影响。
1 实 验
1.1 原材料与碱活化剂
粉煤灰来自河南某电厂,矿渣由华新湘钢水泥公司提供,电石渣来自河南郑州巩义市元亨净水材料厂,原材料的主要化学组成如表1所示。
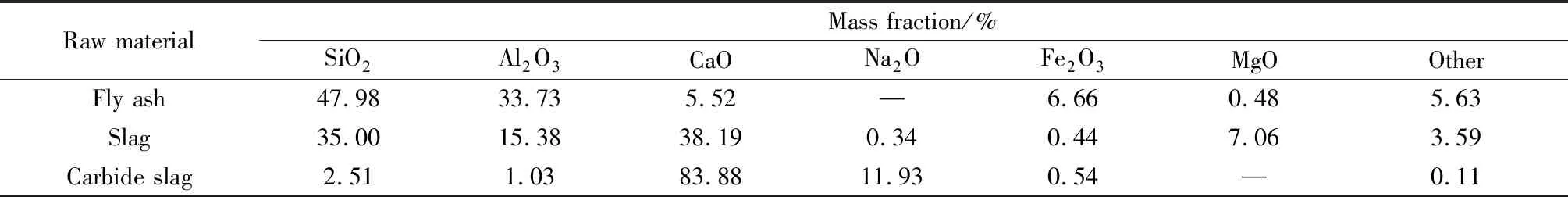
表1 粉煤灰、矿渣和电石渣的主要化学组成
在Na2SiO3溶液中加入固体NaOH和水,配制所需浓度和模数的碱活化剂。Na2SiO3溶液来自河北省邢台市内丘力天化工有限公司,其中Na2O与SiO2的含量分别为8.35%和26.54%(以下均为质量分数),模数M=3.28;固体NaOH来自河南省郑州市清源化工产品有限公司,纯度达99.5%,工业级片状氢氧化钠。
1.2 试验设计
基于前期研究[15],试验设计的配合比如表2所示。
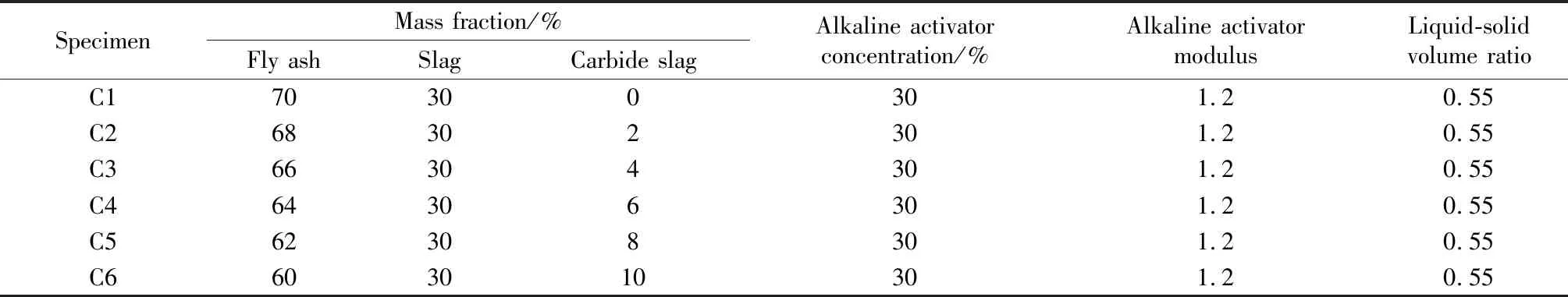
表2 试件配合比设计
在温度为(20±2) ℃、湿度为(95±2)%条件下,标准养护28 d后获得所需试件。将立方体试件的五个面封上石蜡,以未封蜡的面作为碳化面。对试件进行封蜡处理,将封蜡后的试件置于CO2浓度为(20±3)%、相对湿度为(70±5)%、温度为(20±2)℃的碳化箱中。按照《普通混凝土长期性能和耐久性能试验方法标准》(GB/T 50082—2009)进行快速碳化试验,并结合MIP、XRD、TGA和SEM等测试进行微观试验,所用仪器有电位滴定仪(848 Titrino plus)、X射线衍射仪(Bruker D8 Advance)、扫描电子显微镜(蔡司公司EVOMA25)、压汞仪(康塔仪器公司PoreMaster 33)和热重分析仪(PerkinElmer STA 6000)。
1)抗压强度试验:依据《ASTM C109/C109M-20》,测试未碳化和碳化龄期为7、14和28 d试件的抗压强度,试验结果取三个试件的平均值。
2)碳化深度测量:碳化深度测量如图1所示,当碳化时间达到3、7、14、21和28 d时取出试件,沿碳化面垂直方向对试件进行切割,在切割面上喷洒质量分数为1%的酚酞酒精溶液,静置30 s后用游标卡尺取10个点量取碳化深度,取平均值作为该试件的碳化深度。
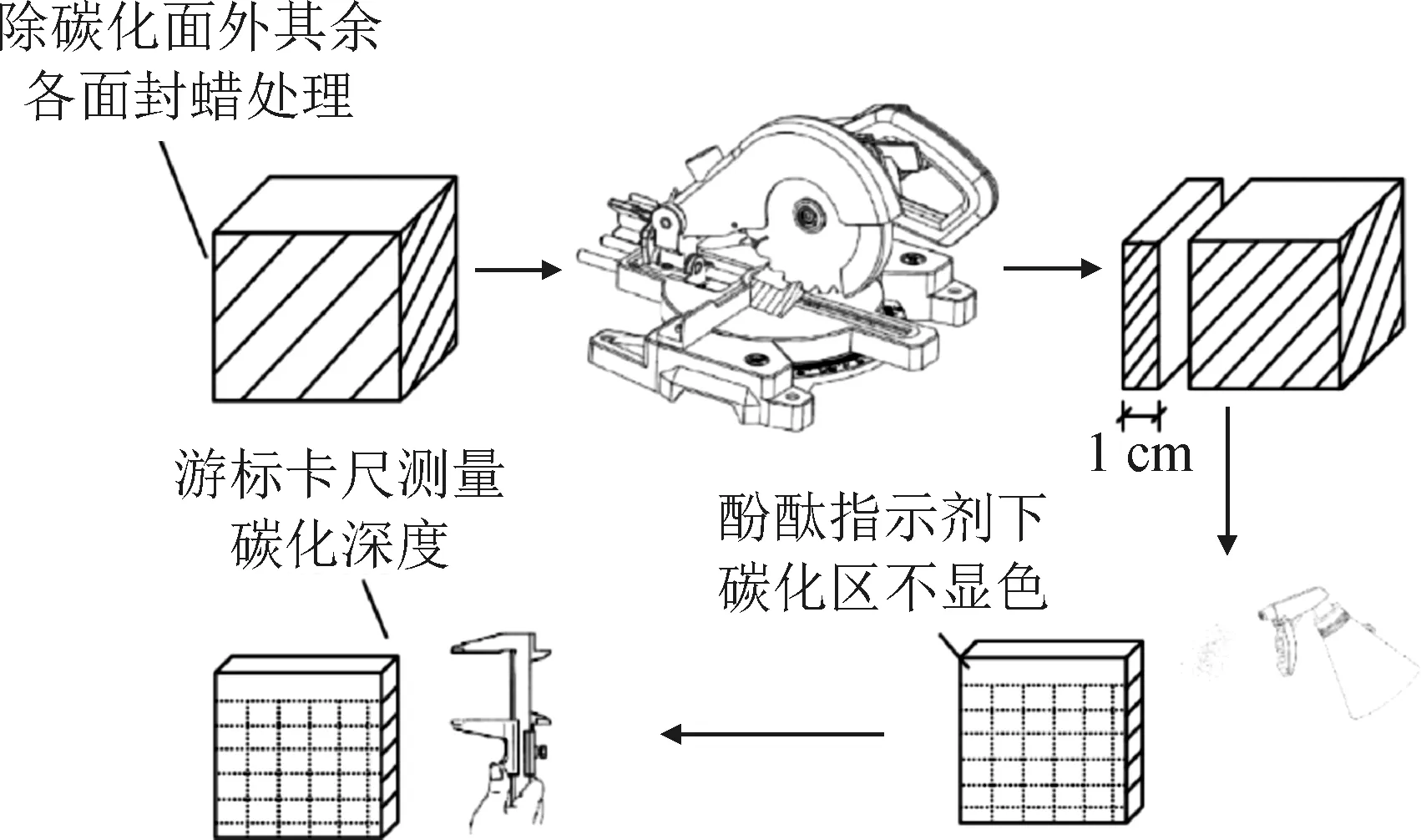
图1 碳化深度测量
3)pH值测试:将试件碳化表面到内部1 cm破碎并研磨成粉,直至全部通过200目(75 μm)筛后再进行烘干。试件粉末与去离子水以摩尔比1∶3混合,混合充分后用电位滴定仪测量上层清液的 pH 值。
4)微观试验:取碳化28 d后的试件,将碳化表面到内部0~10 mm破碎后,选取1 cm3左右的规则碎片,在50 ℃的烘箱中烘干至恒重,通过MIP分析孔结构特征。在碎片平整的断面上喷金,进行SEM测试,观察试件微观形貌。将破碎后的试件烘干至恒重,研磨成粒径小于75 μm的粉末,进行TGA和XRD测试,分析碳化产物。
2 结果与讨论
2.1 碳化深度与孔隙结构分析
图2为不同电石渣掺量下试件的碳化深度。本文以试件总孔隙率乘以孔径后大于50 nm的有害孔隙的权重作为大孔径占比,不同电石渣掺量下试件碳化前的孔隙特征如图3所示。由图2可知,当快速碳化3 d 时,C1的碳化深度为2.24 mm,随着电石渣掺量的增加,碳化深度逐渐降低,当电石渣掺量为10%时C6的碳化深度为0.62 mm。这是因为随电石渣掺量的增加,电石渣带来的Ca(OH)2吸收了CO2,从而改善了CO2从碱激发凝胶材料表层向内部入侵的情况。当快速碳化7、14 d时,随电石渣掺量增加,碱激发凝胶材料的碳化深度仍逐渐减小,但各试件之间的碳化深度差异逐渐变小,C1与C6的碳化深度之差在快速碳化7 d后仅为1.54 mm,14 d后为1.08 mm。由图3可知,电石渣的掺入可增大碱激发凝胶材料的孔隙率,而较高的初始孔隙率会加快CO2在内部的扩散。因此,随着碳化龄期增长,由电石渣提供碱性储备带来的抗碳化性能优势逐渐降低。随着快速碳化时间继续增长,当快速碳化21 d时,部分试件中由电石渣提供的Ca(OH)2可能已与CO2反应完全,因此由孔隙率带来的影响开始成为主导。

图2 不同电石渣掺量下试件的碳化深度
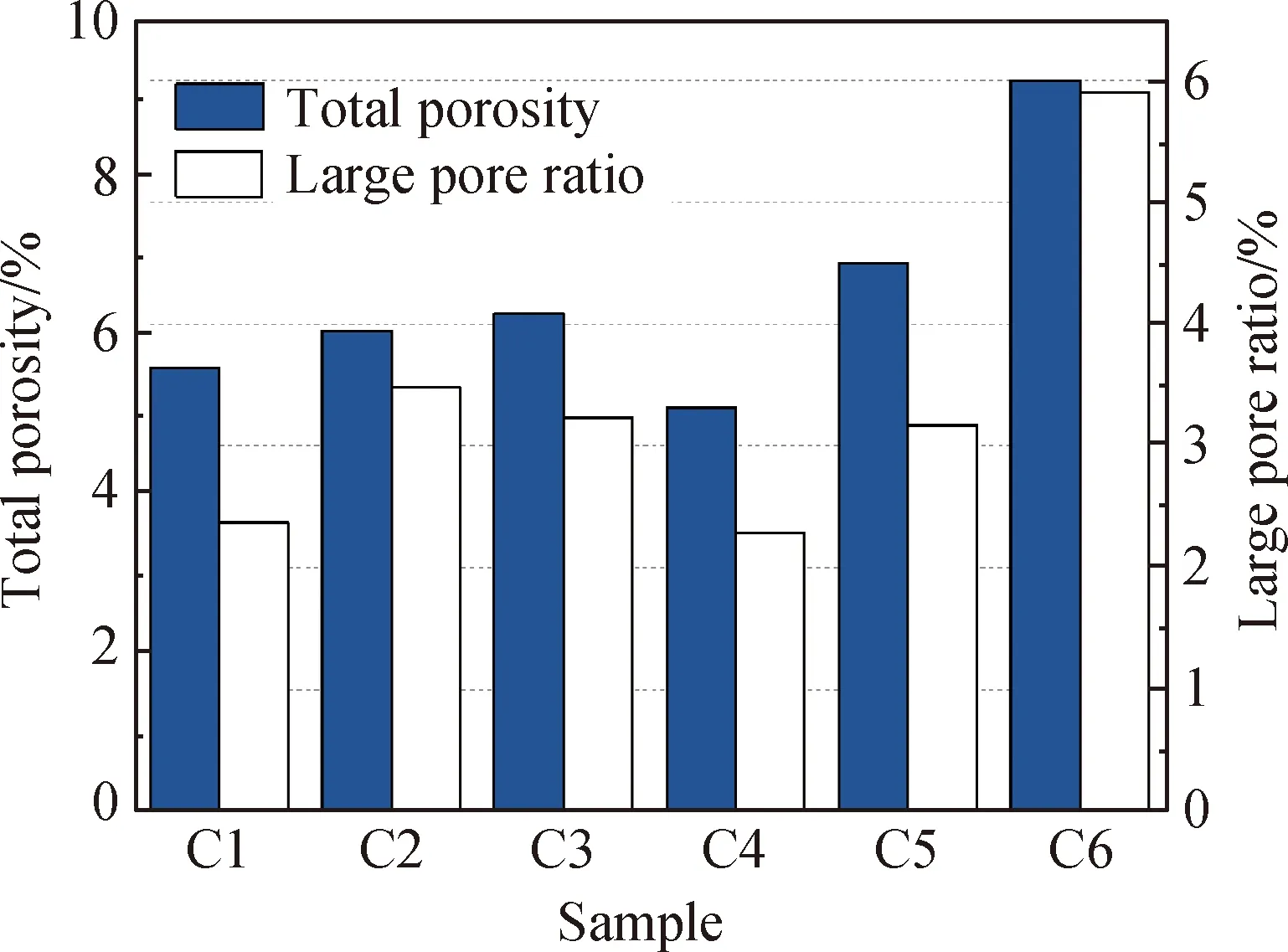
图3 不同电石渣掺量下试件碳化前的孔隙特征
图4为碳化前后试件的总孔隙率、大孔径占比和孔径分布变化。由图4(a)和(b)可知,碳化后试件的总孔隙率和大孔径占比均大于碳化前,这是因为矿渣生成的C-(A)-S-H凝胶发生脱钙,导致孔隙粗化[5],反应生成的碳酸盐并不能有效地填充孔隙。由图4(c)可知,试件经28 d加速碳化作用后,大于1 000 nm的孔径占比均增大,小于50 nm的孔径占比均减小。
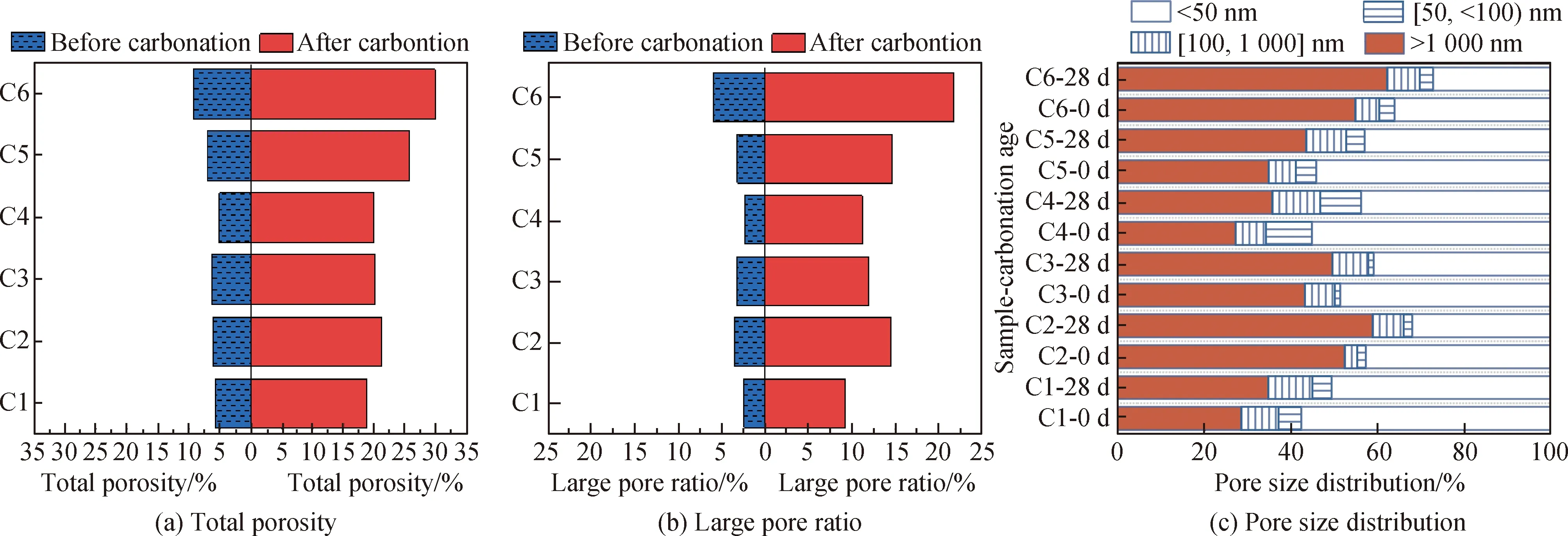
图4 碳化前后试件孔隙特征的变化
C5和C6在快速碳化28 d后总孔隙率和大孔径占比较碳化前的增加量均大于其他组,这说明在碳化作用下,电石渣掺量较高的试件孔隙粗化现象更为严重。这可能是因为在加速碳化后,C5和C6中的Ca(OH)2被消耗,生成的CaCO3不能很好地填充Ca(OH)2留下的孔隙,导致孔隙增大。因此在快速碳化28 d后,随电石渣掺量增加,试件碳化深度的增长幅度逐渐增大,含电石渣的试件(C2~C6) 28 d碳化深度均大于不含电石渣的试件(C1)。
综上分析得知,在碳化早期,电石渣的掺入增加了碱激发凝胶材料的碱度,电石渣含量越高的试件其碳化深度越小。随着碳化时间的推移,由于除电石渣掺量为6%的试件之外,其他含电石渣的试件初始孔隙率相对较高,在碳化过程中孔隙率粗化更为严重,加速了CO2在内部的扩散。因此在碳化的中后期,电石渣对体系碱度的提升逐渐无法补足因孔隙率增大而带来的负面影响,掺入电石渣的试件碳化深度大于不含电石渣的试件,其中C6组的碳化深度大于其他组,碳化深度最高达19.35 mm,失去了抗碳化性能优势。由于加速碳化28 d的碳化深度与50年自然碳化深度基本接近[16],因此在较长的时间内,含电石渣试件的碳化深度发展慢于不含电石渣试件,其中当电石渣掺量为6%时,C4在碳化前中期的碳化深度发展速度最慢。
2.2 碱度变化
电石渣的化学组成中Ca(OH)2含量丰富,掺入后对体系的碱度有一定影响。本文以取代粉煤灰掺量的方式掺入电石渣,而粉煤灰掺量的减少对体系碱度的影响可以忽略不计,因此在分析不同电石渣取代粉煤灰掺量下试件碳化过程时表层pH值的变化前,有必要先研究电石渣掺入对体系中Ca(OH)2含量的影响。本文以改良蔗糖滴定法[17]测定碳化前试件中Ca(OH)2的含量,结果如图5所示。由图5可以直观看出,随电石渣掺量增加,试件中Ca(OH)2的质量分数从2.21%持续上升至8.46%。这证实了电石渣的掺入能使试件在碳化前拥有较高的Ca(OH)2含量,对于减小二氧化碳的入侵速率有积极作用。
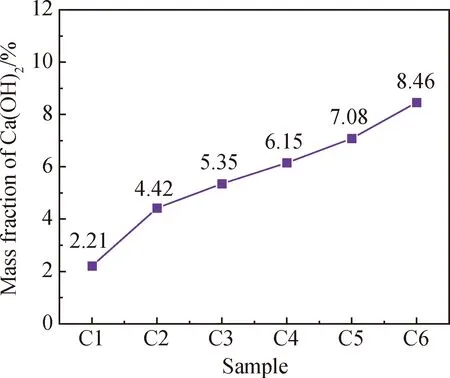
图5 碳化前试件中Ca(OH)2的质量分数
取试件各碳化龄期下离碳化表面1 cm深度的试件粉末进行碱度测试,得到的表层pH值变化如图6所示。由上文可知,碳化前电石渣掺量越大,试件中Ca(OH)2含量越高,因此观察到试件在碳化0 d时pH值随电石渣掺量的增加而逐渐增大。在整个碳化过程中,未掺入电石渣的C1表层pH值从11.65下降至10.45。对于掺入电石渣的试件,由于电石渣的掺入提供了Ca(OH)2,因此在各碳化龄期,电石渣含量越高,试件表层的pH值更高。电石渣提供的Ca(OH)2可以中和部分碳酸,一定程度上维持了体系的碱度,因此含电石渣的试件在碳化3 d内pH值下降缓慢。但随着碱碳化龄期的增长,试件内部形成一定数量的大孔,导致孔隙率增大,CO2的侵入速度随之加快,试件表层pH值的下降速度增快,在28 d碳化结束后,各组试件表层pH值大致分布在两个区间:10.45~10.49(C1~C3)和10.65~10.68(C4~C6)。综上,从试件碳化后的表层pH值来看电石渣的掺入在碳化前中期能为碱激发凝胶材料提供较为有效的碱度保障;在碳化后期,由于受到孔隙率等因素的影响,各电石渣掺量下试件表层的pH值发生了断层,即电石渣掺量小于6%时地聚物表层pH值较低,电石渣掺量大于等于6%时地聚物表层pH值较高,两区间内各试件的pH值差异甚微。
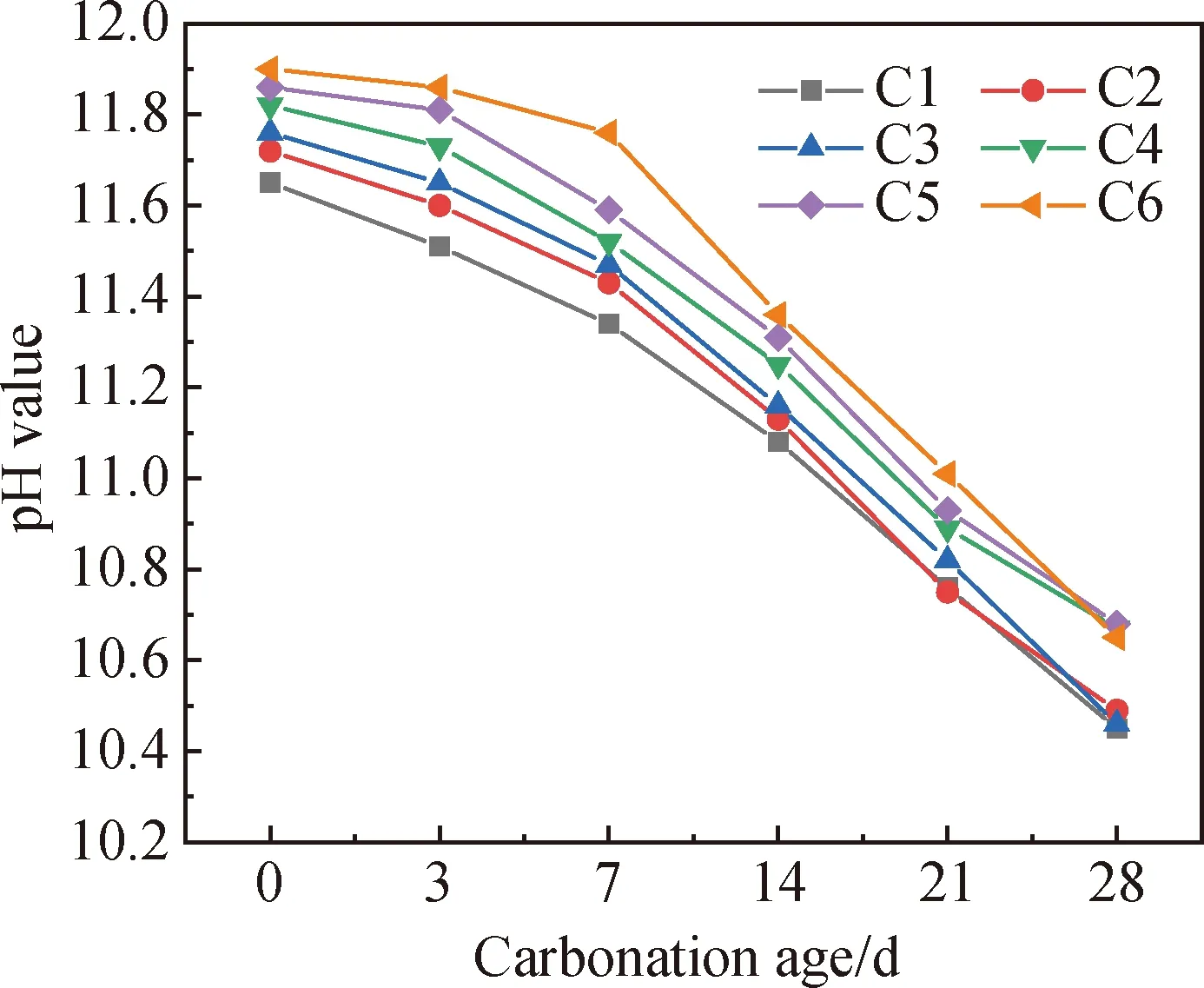
图6 不同碳化龄期下试件表层pH值变化
2.3 抗压强度变化
图7为不同碳化龄期下试件的抗压强度,图8为碳化后试件的抗压强度衰减率。由图7和图8可知,碳化后试件的孔隙结构向大孔径发展,其总孔隙率和大孔径的占比均增大,各组试件碳化后的抗压强度均出现衰减。
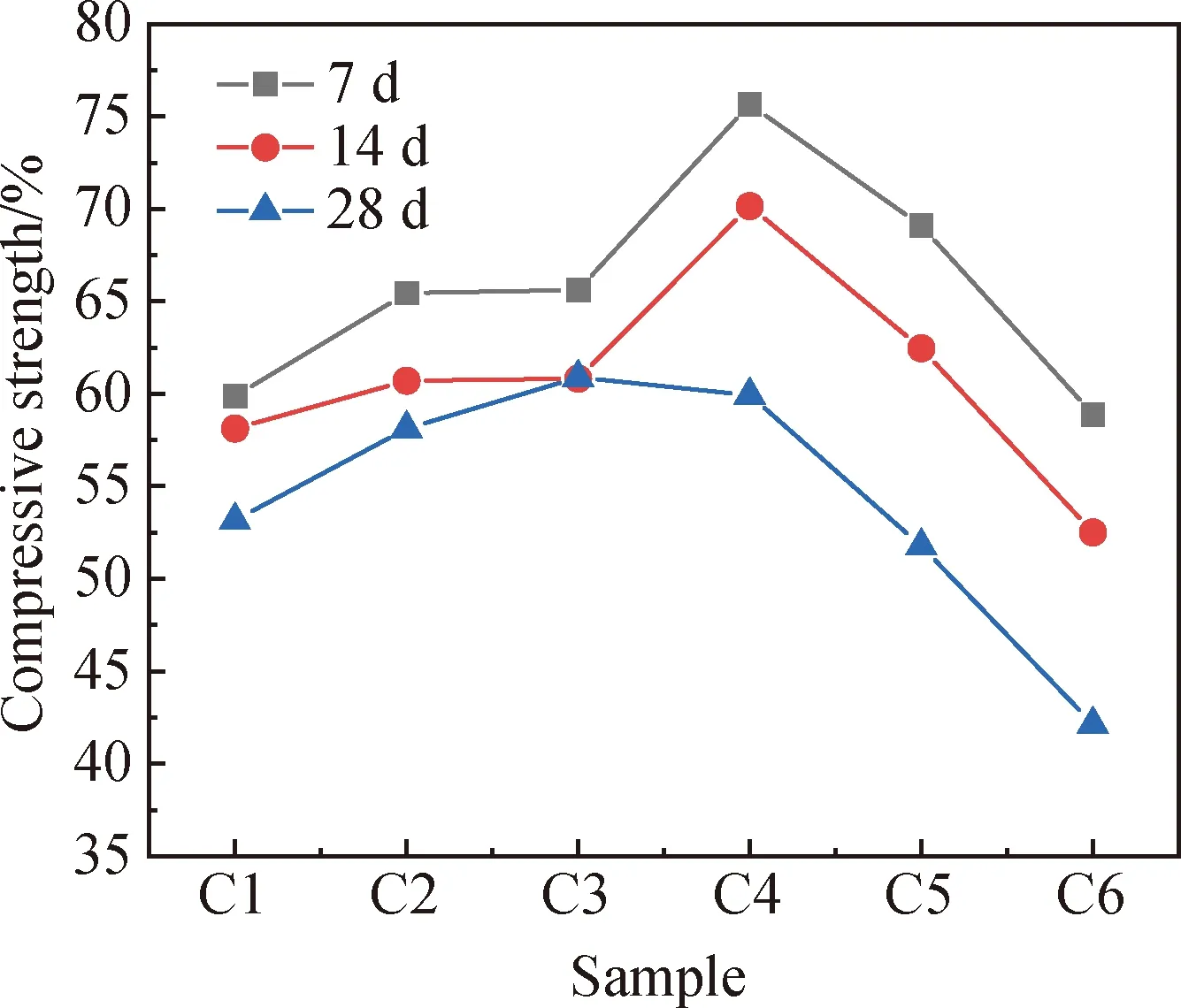
图7 不同碳化龄期下试件的抗压强度

图8 碳化后试件的抗压强度衰减率
当快速碳化7 d时,随电石渣掺量增加,试件的抗压强度衰减率持续降低,其中C1与C6相差12.35个百分点。随着碳化龄期增长至14 d,C4的抗压强度衰减率最低,其余各组无明显规律。当碳化龄期为28 d时,含电石渣的试件(C2~C6)碳化后抗压强度衰减率均大于不含电石渣的试件(C1)。且当电石渣掺量大于6%之后,试件的抗压强度衰减率明显增加,其中C6碳化后的抗压强度衰减率较C1高14.4个百分点。然而,电石渣掺量为2%~6%的试件在28 d碳化后的抗压强度仍高于C1。说明电石渣的掺入虽然有使碱激发凝胶材料的孔隙在碳化后向有害孔隙发展的趋势,但在适当的电石渣掺量下,仍能获得高于碳化后C1的抗压强度。
由上文可知,电石渣的掺入能在碳化前期降低碱激发凝胶材料的抗压强度衰减率,随着碳化龄期推移至28 d,掺入电石渣的碱激发凝胶材料已无抗压强度衰减率的优势。但在合适的电石渣掺量(6%)下,碱激发凝胶材料仍能获得较高的碳化后抗压强度。综上,结合碳化深度发展、pH值降低和抗压强度衰减率来看,电石渣掺量为6%的试件在碳化前中期具备最佳抗碳化性能,并且在碳化后期仍拥有最大抗压强度,为39.92 MPa。
图9为28 d碳化前后试件大孔径占比差值与抗压强度衰减率关系。由图9可知,碱激发凝胶材料作为一种常见的多孔性材料,其碳化前后大孔径占比差值与抗压强度衰减率之间存在着较为明显的正相关关系。这意味着,随着试件碳化前后大孔径占比差值的增大,其碳化后的抗压强度衰减率也会相应增大。这是因为碳化过程会使碱激发凝胶材料产生内部应力和孔隙率变化,大孔径对碳化后的结构稳定性具有更大的影响,从而影响其抗压强度的衰减情况。

图9 28 d碳化前后试件大孔径占比差值与抗压强度衰减率关系
2.4 碳化产物分析
对C1-0 d(碳化 0 d)、C4-0 d(碳化 0 d)和C4-28 d(碳化 28 d)的试件进行热重分析,碳化前后试件的TG-DTG曲线如图10所示。在C1-0 d的DTG曲线中,69.08和130.56 ℃的峰值分别代表了基体中自由水的蒸发和水化硅酸钙(C-S-H)凝胶中结合水的损失[18-21]。根据Nedeljkovi等[22]的研究可知,无定形CaCO3的特定分解温度范围在245~645 ℃,因此认为本文中276.20 ℃处的峰值与无定形碳酸盐分解、C-A-S-H和C-A-H基质脱水有关[2,18-19]。Ca(OH)2分解的峰值通常出现在450 ℃附近[18,20],460.59 ℃的峰值主要由Ca(OH)2和无定形CaCO3的分解所致。C1-0 d的DTG曲线在638.77 ℃处出现峰值,这是因为CaCO3组分的分解[2,19]或代表球霰石和方解石的特征峰[20-21]。曲线在786.08 ℃处的峰值则与结晶CaCO3的分解有关[2,19]。

图10 碳化前后试件的TG-DTG曲线
由图10中C4-0 d的DTG曲线可知,曲线出现峰值的位置几乎与C1-0 d相吻合,说明了含电石渣试件与不含电石渣试件的物质组成相似。但对比C1-0 d与C4-0 d的DTG曲线发现,后者的DTG曲线在460.59 ℃出现的峰值大于前者在454.52 ℃出现的峰值,这很可能与两者的Ca(OH)2含量差异有关。上文通过蔗糖滴定法测得C4中Ca(OH)2质量分数为6.15%,C1中Ca(OH)2为2.21%,进一步证实了此猜想的可能性。
对比图10中C4-0 d与C4-28 d的热重分析发现,在碳化前后的DTG曲线中,峰值出现的位置仍大致相同。对比两者的DTG曲线发现,C4-28 d的DTG失重峰位置出现在了428.15 ℃处,相对于碳化前的试件前移了32.44 ℃,且峰值下降明显。这是因为碳化过程消耗了Ca(OH)2,碳化后的试件可能已不含Ca(OH)2或含量很少,因此可以猜想此处的失重峰主要为无定形碳酸钙的分解。C4-28 d的DTG曲线在622.90 ℃处的峰值略低于C4-0 d在643.10 ℃处的峰值,但在796.92 ℃处的峰值大于C4-0 d在757.62 ℃的峰值,这可能是由于碳化过程生成了结晶较为良好的碳酸盐。
对快速碳化28 d的C4试件进行晶相组成分析,并标记部分主要的晶相,得到的XRD谱如图11所示,含电石渣试件的碳化产物中未发现Ca(OH)2晶体,但观察到方解石、霰石等碳酸盐晶体,其中方解石对应了热重分析中622.90 ℃处的失重峰。这进一步验证了AAFSC在快速碳化28 d后,碳化区的Ca(OH)2被消耗,且生成了更多CaCO3等碳酸盐晶体。

图11 碳化28 d后C4试件的XRD谱
在碳化龄期为0和28 d的C4试件进行SEM观测,得到的SEM照片如图12所示。普通混凝土和粉煤灰基地聚物混凝土经过碳化作用后,生成的CaCO3填充孔隙,结构更加致密,这与含电石渣试件相反。对比图12(a)和(b)发现,碳化前致密的凝胶在碳化作用后变得疏松多孔,存在明显的凝胶脱钙现象。除此之外,从图12中还可观察到许多不同形状的颗粒,其中球状颗粒为未反应的粉煤灰或球形CaCO3等。

图12 碳化前后C4试件的SEM照片
3 结 论
1)经过快速碳化作用,含电石渣试件的孔隙结构会向有害孔发展,抗压强度存在明显衰减,且碳化后大孔径占比的增值与抗压强度衰减率呈正相关。
2)适当掺入电石渣有利于在碳化前期减小碱激发粉煤灰-矿渣的碳化速率,但随碳化龄期延长,这种优势逐渐减小甚至失去;结合碳化深度发展、pH值降低和抗压强度衰减来看,电石渣掺量为6%的试件在碳化前中期具备最佳抗碳化性能,并且在碳化后期仍拥有最大抗压强度39.92 MPa。
3)随电石渣掺量增加,含电石渣试件中Ca(OH)2含量增加,这些Ca(OH)2在碳化过程中被消耗,生成了方解石、霰石等碳酸盐。
