消渴健脾胶囊制备工艺、质量标准研究
2023-09-19丁然然杨明霞何承辉马美玲
丁然然,杨明霞,何承辉,马美玲
(1.新疆医科大学,新疆 乌鲁木齐 830054;2.乌鲁木齐市中医医院,新疆 乌鲁木齐 830099;3.新疆维吾尔自治区药物研究所,新疆 乌鲁木齐 830004)
消渴健脾胶囊由苍术、白术等13 味中药组成,具有行气和胃、燥湿健脾等功效,为乌鲁木齐市中医医院国家级名老中医许公平主任的临床经验方,至今已有二十余年临床应用历史,用于治疗脾虚湿盛型2 型糖尿病[1-2],可调节糖尿病患者炎性因子,减轻胰岛素抵抗,改善形体肥胖、身体困倦、头胀、脉沉等脾虚湿盛型证候,提高生活质量[3-5]。为了满足医院临床需要,本实验以中医药理论为指导,根据国家食品药品监督管理总局关于对医疗机构应用传统工艺配置中药制剂实施备案管理公告(2018 年第19 号) 的要求,开展消渴健脾胶囊临床前药学研究工作。另外,课题组前期曾将制备工艺更改为部分药材水提取,部分药材乙醇提取,部分药材提取挥发油,但专家认为消渴健脾胶囊原工艺已有多年临床实践支撑,疗效显著,安全可靠,建议维持原工艺,故本实验在原制备工艺基础上进行[6-7]。
查阅文献[8-12] 发现,消渴健脾胶囊中绿原酸、葛根素、大豆苷、毛蕊花糖苷、落新妇苷、丹酚酸B、大豆苷元是主要降糖活性成分,并且含量较高,常作为质量控制指标。因此,本实验建立HPLC 法同时测定上述成分的含量,以期控制消渴健脾胶囊中茵陈、葛根、车前子、土茯苓、丹参质量,并为该制剂质量标准研究提供参考。
1 材料
1.1 仪器 Agilent 1260、Agilent 1200 型高效液相色谱仪(美国Agilent 公司);KQ-500DE 型超声波清洗机(郑州长城科工贸有限公司);WP-UP-IV-10 Water Purifier 型实验室专用超纯水机(四川沃特尔科技发展有限公司);DZF-6090 型真空干燥箱(上海齐欣科学仪器有限公司)。
1.2 药材、对照品 茯苓、栀子均购自新疆众安康中药饮片有限公司(批号20200802、20200403);木瓜、丹参、茵陈均购自安徽济善堂中药科技有限公司(批号200801、200901、200501);苍术、大黄均购自安徽人民中药饮片有限公司(批号190701、190801);莱菔子、厚朴均购自新疆鸿德堂永盛药业有限公司(批号200601、200901);葛根、白术均购自安徽家和中药科技股份有限公司 (批号191104、200601);车前子购自毫州市华云中药饮片有限公司(批号190601);土茯苓购自安国市祁澳中药饮片有限公司(批号2007839113),经新疆医科大学田树革教授鉴定为正品。落新妇苷对照品购自上海中药标准化研究中心(批号30-2017);栀子苷对照品、栀子对照药材、大黄酚对照品、大黄对照药材、丹参对照药材、葛根素对照品均购自中国食品药品检定研究院(批号120986-201610、120986-201510、110758-201507、120902-201311、121117-201608、110752-201813);大豆苷、大豆苷元、毛蕊花糖苷、丹酚酸B 、绿原酸对照品均购自四川省维克奇生物科技有限公司 (批号wkq21041310、wkq21120208、wkq20042004、wkq 21010705、wkq20081302)。
1.3 试剂与药物 消渴健脾胶囊 (批号20210103,纯度82%) 由乌鲁木齐市中医医院药剂科提供。甲醇、乙腈为色谱纯;其他试剂均为分析纯;水为自制超纯水。
2 制备工艺研究
2.1 落新妇苷含量测定 采用HPLC 法。
2.1.1 色谱条件 COSMOSIL packed Column 5C18-MS-Ⅱ色谱柱(4.6 mm×250 mm,5 μm);流动相甲醇-0.1% 冰醋酸 (37 ∶ 63);体积流量1.0 mL/min;柱温30 ℃;检测波长291 nm。理论塔板数按落新妇苷峰计算,应不低于5 000[13]。色谱图见图1。
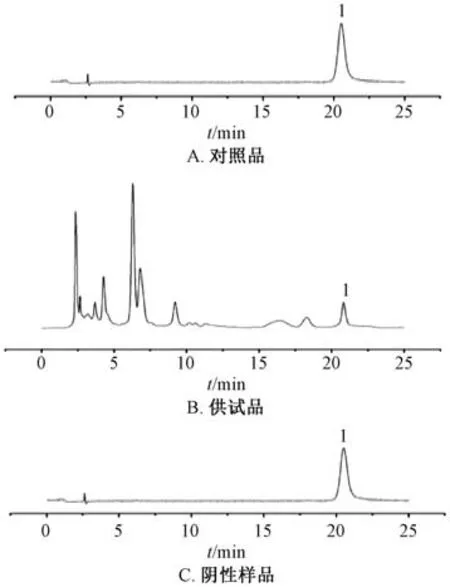
图1 落新妇苷HPLC 色谱图Fig.1 HPLC chromatograms of astilbin
2.1.2 对照品溶液制备 取落新妇苷对照品适量,甲醇溶解,摇匀,稀释至刻度,即得。
2.1.3 供试品溶液制备 按本品处方比例,称取苍术、茵陈、栀子、丹参、土茯苓、莱菔子、厚朴、木瓜适量,置于2 000 mL 量瓶中,加10 倍量水,置于调温电热套中加热回流提取1 h,提取液过滤后置于2 000 mL 量瓶中,滤渣加8 倍量水提取45 min,提取液过滤后置于2 000 L量瓶中,合并提取液,加水定容至刻度,上清液用0.22 μm微孔滤膜过滤,取续滤液,即得。
2.1.4 阴性样品溶液制备 按本品处方比例,制成缺土茯苓的阴性样品,按“2.1.3” 项下方法制备,即得。
2.2 栀子苷含量测定 采用HPLC 法。
2.2.1 色谱条件 COSMOSIL packed Column 5C18-MS-Ⅱ色谱柱(4.6 mm×250 mm,5 μm);流动相乙腈-水(13 ∶87);体积流量1.0 mL/min;柱温30 ℃;检测波长238 nm。理论塔板数按栀子苷峰计,应不低于1 500[14]。色谱图见图2。
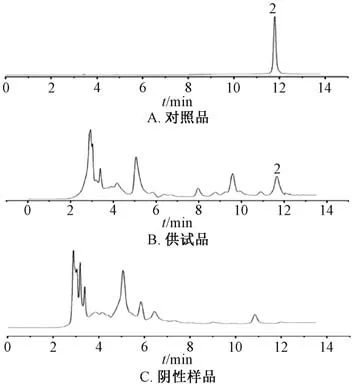
图2 栀子苷HPLC 色谱图Fig.2 HPLC chromatograms of gardenoside
2.2.2 对照品溶液制备 取栀子苷对照品适量,甲醇溶解,摇匀,稀释至刻度,即得。
2.2.3 供试品溶液制备 取“2.1.3” 项下提取液适量,上清液用0.22 μm 微孔滤膜过滤,取续滤液,即得。
2.2.4 阴性样品溶液制备 按本品处方比例,制成缺栀子的阴性样品,按“2.1.3” 项下方法制备,即得。
2.3 水浸出物含量测定 精密吸取提取液50 mL,置于干燥至恒定质量的蒸发皿中,水浴蒸干,置于105 ℃烘箱中干燥3 h 至恒定质量,取出,迅速置于干燥器中,冷却,称定质量,计算含量。
2.4 单因素试验 以加水量、提取时间、提取次数为影响因素,栀子苷、落新妇苷、水浸出物含量为评价指标,最大权重分别设定为40、40、20,计算综合评分,公式为综合评分=(栀子苷含量/最大栀子苷含量) ×40+ (落新妇苷含量/最大落新妇苷含量) ×40+ (水浸出物含量/最大水浸出物含量) ×20。
2.4.1 加水量 按本品处方比例,称取苍术、茵陈、栀子、丹参、土茯苓、莱菔子、厚朴、木瓜适量,平行4 份,分别加入8、10、12、14 倍量水各回流提取2 次,每次1 h,提取液过滤后置于2 000 mL 量瓶中,加水定容至刻度,摇匀,吸取适量,过0.22 μm 微孔滤膜,取续滤液,测定落新妇苷、栀子苷、水浸出物含量,结果见表1。由此可知,综合评分随着加水量增加呈升高趋势,为8 倍量以上时大于90 分,故选择8、10、12 倍量水进行正交试验。
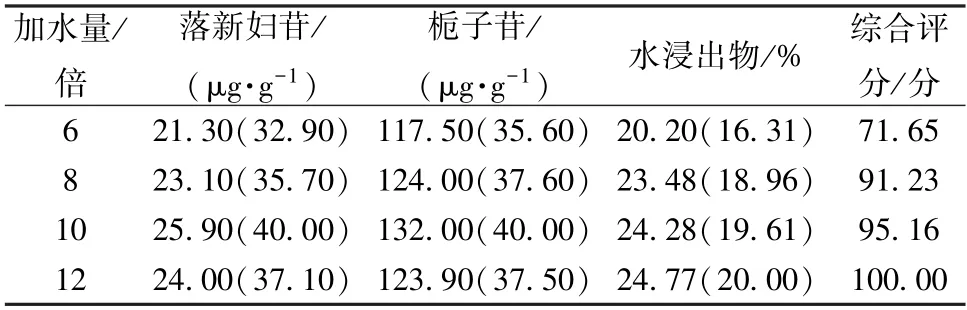
表1 加水量单因素试验结果Tab.1 Results of single factor tests for water addition
2.4.2 提取时间 按本品处方比例,称取苍术、茵陈、栀子、丹参、土茯苓、莱菔子、厚朴、木瓜适量,平行4 份,加10 倍量水分别提取0.5、1、1.5、2 h 各2 次,提取液合并后置于2 000 mL 量瓶中,加水定容至刻度,摇匀,取上清液,过0.22 μm 微孔滤膜,取续滤液,测定落新妇苷、栀子苷、水浸出物含量,结果见表2。由此可知,综合评分随着提取时间延长先升后降,表明时间过长会降低提取效率,故选择0.5、1、1.5 h 进行正交试验。
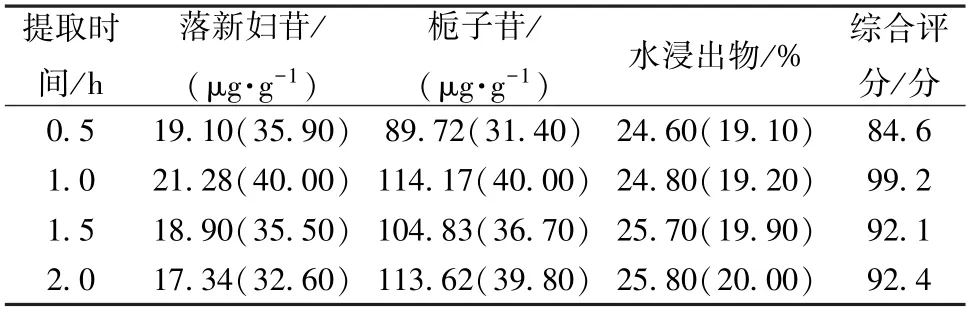
表2 提取时间单因素试验结果Tab.2 Results of single factor tests for extraction time
2.4.3 提取次数 按本品处方比例,称取苍术、茵陈、栀子、丹参、土茯苓、莱菔子、厚朴、木瓜适量,平行4 份,置于圆底烧瓶中,分别编号1、2、3、4,其中1 号样品加1 508 mL 水提取1 次,共2 h;2 号样品加754 mL 水提取2 次,每次1 h;3 号样品第1、2 次各加528 mL 水分别提取1、0.5 h,第3 次加452 mL 水提取0.5 h,提取液过滤后置于2 000 mL 量瓶中,加水定容至刻度,摇匀,取上清液,过0.22 μm 微孔滤膜,取续滤液,测定落新妇苷、栀子苷、水浸出物含量,结果见表3。由此可知,提取2 次时综合评分最高,故选择1、2、3 次进行正交试验。
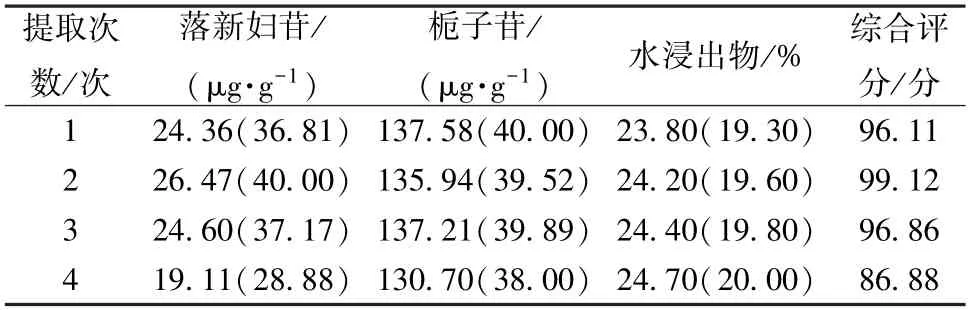
表3 提取次数单因素试验结果Tab.3 Results of single factor tests for extraction frequency
2.5 正交试验 按本品处方比例,称取苍术、茵陈、栀子、丹参、土茯苓、莱菔子、厚朴、木瓜共75.4 g,在单因素试验基础上,以提取次数(A)、加水量(B)、提取时间(C) 为影响因素,栀子苷、落新妇苷、水浸出物含量为评价指标,权重系数分别设定为0.4、0.4、0.2,采用L9(34) 正交表进行设计,因素水平见表4。
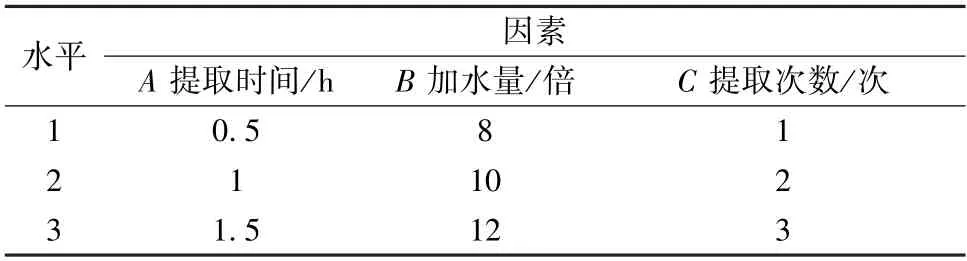
表4 因素水平Tab.4 Factors and levels
按本品处方比例,称取苍术、茵陈、栀子、丹参、土茯苓、莱菔子、厚朴、木瓜适量,以表4 因素水平进行提取,滤过,合并提取液,置于2 000 mL 量瓶中,加水定容至刻度,摇匀,取上清液,过0.22 μm 微孔滤膜,取续滤液,测定落新妇苷、栀子苷、水浸出物含量,结果见表5。
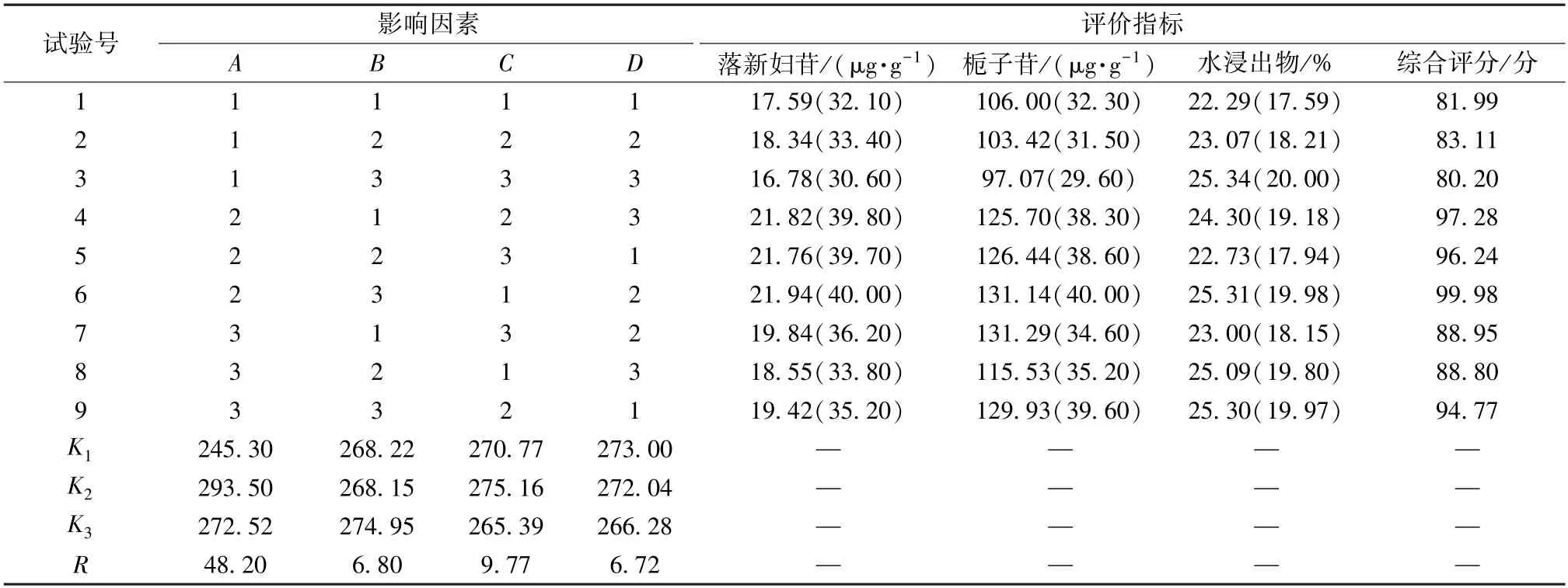
表5 试验设计与结果Tab.5 Design and results of tests
对表5 数据进行方差分析,结果见表6。由此可知,各因素影响程度依次为提取时间>提取次数>加水量;提取时间对提取效果有显著影响(P<0.05),而提取次数、加水量无显著影响(P>0.05)。为了降低成本、节省工时,并结合单因素试验结果,最终确定最优工艺为A2B2C2,即提取时间1.0 h,加水量10 倍,提取次数2 次。
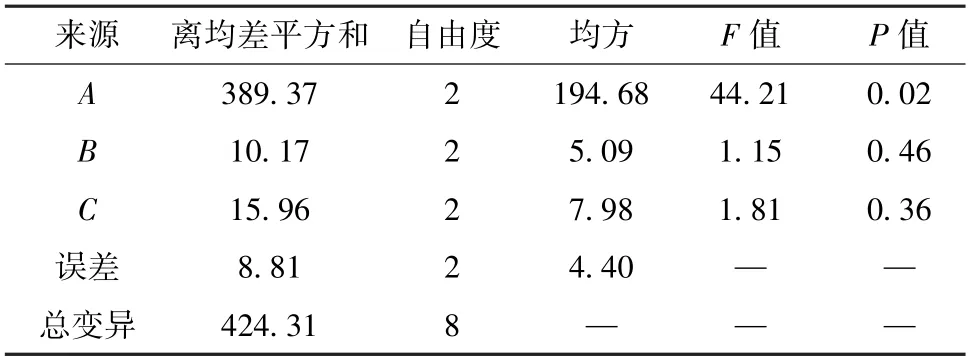
表6 方差分析Tab.6 Analysis of variance
再按上述优化工艺进行3 批验证试验,结果见表7。由此可知,该工艺稳定可行。
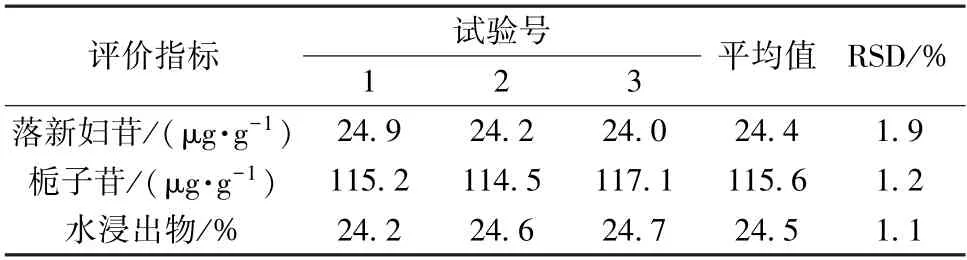
表7 验证试验结果(n=3)Tab.7 Results of verification tests (n=3)
3 质量标准研究
3.1 定性鉴别 采用TLC 法。
3.1.1 栀子、葛根
3.1.1.1 供试品溶液制备 将本品内容物研细,取约3 g,置于具塞锥形瓶中,30 mL 甲醇超声提取45 min,滤过,水浴蒸干滤液,20 mL 水溶解残渣,稀盐酸调pH 至约为2,20 mL 乙醚振摇提取2次,除去乙醚液,水液加20 mL 二氯甲烷振摇提取2 次,除去二氯甲烷液,水液加20 mL 乙酸乙酯振摇提取2 次,合并水液,调pH 至中性,20 mL 乙酸乙酯提取2 次,水液再用水饱和正丁醇提取2次,每次20 mL,合并正丁醇液,水浴蒸干,1 mL甲醇溶解残渣,即得。
3.1.1.2 对照品溶液制备 取栀子苷、葛根素对照品适量,甲醇制成质量浓度为1 mg/mL 的溶液,即得。
3.1.1.3 阴性样品溶液制备 根据本品处方比例,制成缺栀子、缺葛根的阴性样品,按“3.1.1.1”项下方法制备,即得。
3.1.1.4 鉴别方法 对照品、供试品、阴性样品溶液在硅胶G 薄层板上各点样6 μL,以三氯甲烷-甲醇-水(14 ∶5 ∶0.5) 为展开剂,在365 nm 紫外灯下检视,或喷以5%香草醛硫酸溶液,热风加热至斑点清晰,结果见图3。由此可知,供试品在对照品同一位置处均显示相同斑点,阴性无干扰。
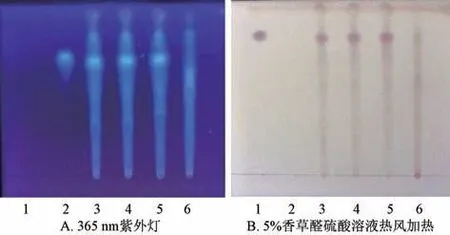
图3 葛根、栀子TLC 色谱图Fig.3 TLC chromatograms of Puerariae lobatae Radix and Gardeniae Fructus
3.1.2 丹参、大黄
3.1.2.1 供试品溶液制备 将本品内容物研细,取约3 g,置于具塞锥形瓶中,40 mL 甲醇溶解,超声处理30 min,滤过,滤液水浴蒸干,20 mL 水溶解残渣,25 mL 乙醚提取,除去乙醚液,水液用盐酸调pH 至约为2~3,25 mL 乙醚提取,除去乙醚液,水液用25 mL 乙酸乙酯提取2 次,合并乙酸乙酯液,30 mL 水洗涤,弃去水液,水浴蒸干,1 mL甲醇溶解残渣,即得。
3.1.2.2 对照药材溶液制备 取丹参对照药材0.5 g,20 mL 盐酸回流提取45 min,滤过,25 mL乙酸乙酯振摇提取2 次,合并乙酸乙酯液,水浴蒸干,1 mL 甲醇溶解残渣,即得相应溶液。再取大黄对照药材0.5 g,20 mL 甲醇溶解,超声提取30 min,取5 mL 滤液,水浴蒸干,20 mL 水+1 mL 盐酸溶解残渣,加热回流提取30 min,冷却至室温,20 mL 乙醚提取2 次,合并提取液,水浴蒸干,2 mL 三氯甲烷溶解残渣,即得相应溶液。
3.1.2.3 阴性样品溶液制备 根据本品处方比例,制成缺丹参、缺大黄的阴性样品,按“3.1.2.1”项下方法制备,即得。
3.1.2.4 鉴别方法 供试品、对照药材、阴性样品溶液在硅胶G 薄层板上各点样3~5 μL,以甲苯-乙酸乙酯-甲酸(8 ∶5 ∶3) 为展开剂,置于氨蒸气中放置15 min,在365 nm 紫外灯下检视,结果见图4。由此可知,供试品在对照药材同一位置处显示相同斑点,阴性无干扰。

图4 大黄、丹参TLC 色谱图Fig.4 TLC chromatogram of Rhei Radix et Rhizoma and Salviae miltiorrhizae Radix et Rhizoma
3.2 含量测定 采用HPLC 法。
3.2.1 色谱条件 Symmetry C18色谱柱(250 mm×4.6 mm,5 μm);流动相甲醇-0.1%磷酸,梯度洗脱,程序[15]见表8;体积流量0.8 mL/min;柱温30 ℃;检测波长294 nm;进样量10 μL。

表8 梯度洗脱程序Tab.8 Gradient elution programs
3.2.2 对照品溶液制备 取绿原酸、葛根素、大豆苷、毛蕊花糖苷、落新妇苷、丹酚酸B、大豆苷元对照品适量,置于5 mL 棕色量瓶中,甲醇超声溶解定容,摇匀,即得(各成分质量浓度分别为0.331、2.392、0.770、1.084、0.208、1.322、0.148 mg/mL)。
3.2.3 供试品溶液制备 将本品内容物研细,取约1 g,置于50 mL 具塞锥形瓶中,20 mL 75%甲醇溶解,称定质量,超声(250 W、40 kHz) 提取45 min,冷却至室温,75% 甲醇补足减失的质量,滤过,取续滤液,0.22 μm 微孔滤膜过滤,取续滤液,即得。
3.2.4 阴性样品溶液制备 根据本品处方比例,制成缺茵陈、葛根、土茯苓、车前子、丹参的阴性样品,按“3.2.3” 项下方法制备,即得。
3.2.5 方法学考察
3.2.5.1 专属性试验 取对照品、供试品、阴性样品溶液适量,在“3.2.1” 项色谱条件下进样测定,结果见图5。由此可知,各成分色谱峰均可明显分离(分离度>1.5),理论塔板数均符合2020年版《中国药典》 单味药材含量测定标准,阴性无干扰,表明该方法专属性良好。
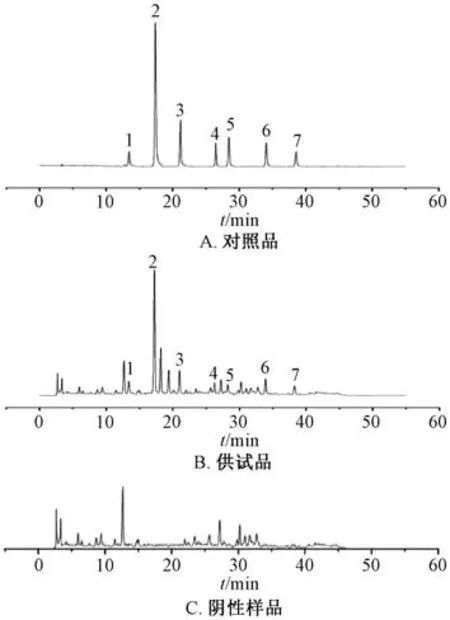
图5 各成分HPLC 色谱图Fig.5 HPLC chromatograms of various constituents
3.2.5.2 线性关系考察 精密吸取“3.2.2” 项下对照品溶液100、250、500、750、1 000 μL,置于5 mL 棕色量瓶中,75%甲醇定容,得系列质量浓度,在“3.2.1” 项色谱条件下进样测定。以对照品质量浓度为横坐标 (X),峰面积为纵坐标(Y) 进行回归,并分别以信噪比3 ∶1、10 ∶1 为检测下限、定量下限,结果见表9,可知各成分在各自范围内线性关系良好。
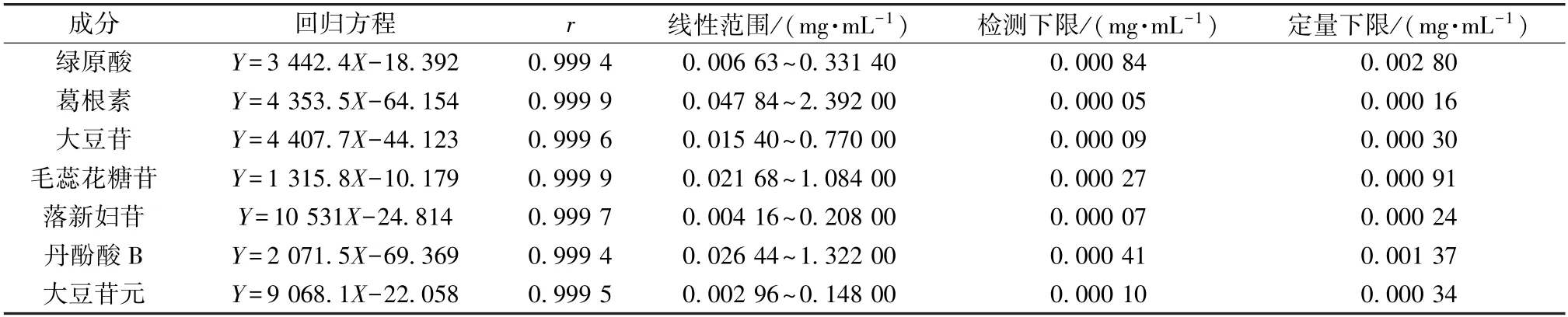
表9 各成分线性关系Tab.9 Linear relationships of various constituents
3.2.5.3 精密度试验 取同一份对照品溶液,在“3.1.2” 项色谱条件下进样测定6 次,测得绿原酸、葛根素、大豆苷、毛蕊花糖苷、落新妇苷、丹酚酸B、大豆苷元峰面积RSD 分别为0.51%、1.70%、0.07%、0.50%、0.57%、0.73%、0.55%,表明仪器精密度良好。
3.2.5.4 稳定性试验 取同一份供试品溶液,室温下于0、2、4、6、8、12 h 在“3.2.1” 项色谱条件下进样测定,测得绿原酸、葛根素、大豆苷、毛蕊花糖苷、落新妇苷、丹酚酸B、大豆苷元峰面积RSD 分别为1.45%、1.15%、1.14%、1.77%、1.82%、1.53%、0.80%,表明溶液在12 h 内稳定性良好。
3.2.5.5 重复性试验 取同一批本品,按“3.2.3” 项下方法平行制备6 份供试品溶液,在“3.2.1” 项色谱条件下进样测定,测得绿原酸、葛根素、大豆苷、毛蕊花糖苷、落新妇苷、丹酚酸B、大豆苷元峰面积RSD 分别为1.44%、1.73%、2.00%、2.03%、1.72%、1.75%、1.01%,表明该方法重复性良好。
3.2.5.6 加样回收率试验 取各成分含量已知的本品粉末6 份,加入对照品适量,按“3.2.3” 项下方法制备供试品溶液,在“3.2.1” 项色谱条件下进样测定,计算回收率。结果,绿原酸、葛根素、大豆苷、毛蕊花糖苷、落新妇苷、丹酚酸B、大豆苷元平均加样回收率分别为98.0%、96.9%、97.7%、100.5%、99.3%、99.5%、98.4%,RSD分别为1.52%、1.21%、0.82%、2.62%、1.17%、1.76%、1.49%。
3.2.5.7 样品含量测定 取3 批中试样品(批号20201101、20201202、20210103),每批3 份,按“3.2.3” 项下方法制备供试品溶液,在“3.2.1”项色谱条件下进样测定,计算含量,结果见表10。
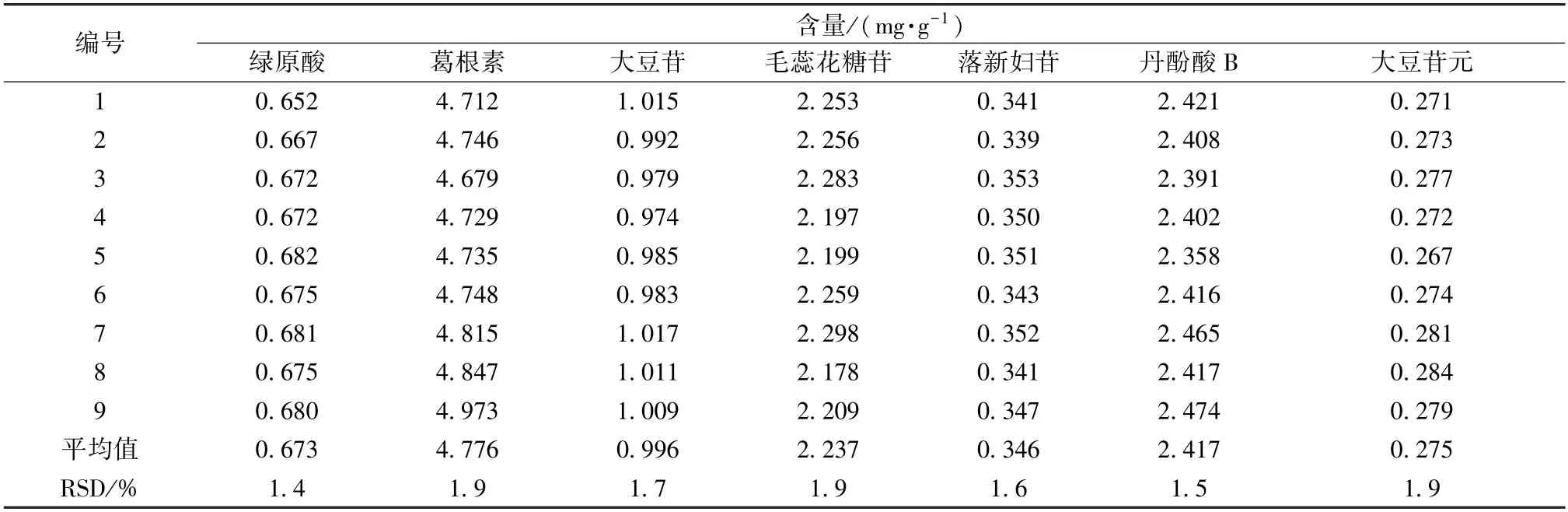
表10 各成分含量测定结果Tab.10 Results of content determination of various constituents
4 讨论
4.1 评价指标选择 君药苍术指标成分苍术素为挥发性化合物,难溶于水,含量较低,故选择臣药栀子、土茯苓中的主要成分。2020 年版《中国药典》 一部规定,栀子中栀子苷含量不得低于1.8%,土茯苓中落新妇苷含量不得低于0.45%,两者不仅是主要降糖活性成分,而且含量较高,易溶于水,化学性质稳定,测定方法成熟,同时水浸出物含量能侧面反应苍术等8 味药材提取率。结合文献[16-18],本实验以栀子苷、落新妇苷、水浸出物含量为评价指标。
4.2 TLC 鉴别方法和药材选择 本实验鉴别栀子、葛根时,参照2020 年版《中国药典》 中牛黄净脑片制剂项下方法;鉴别大黄、丹参时,以对照药材定性,供试品溶液中与大黄对应的点为大黄酚,而与丹参对应的点尚不明确,有待进一步探索。同时,栀子、葛根选择同一处理方法、展开系统及不同显色方法进行鉴别,而大黄、丹参选择同一处理方法、展开条件、显色方法进行鉴别,既可简化操作步骤,又能节能减排。另外,白术、茯苓、车前子、木瓜、茵陈均有阴性干扰,经检测方法筛选、展开系统调整后仍无法排除;厚朴虽然无阴性干扰,但目标斑点模糊,不易辨认,故本实验未对上述6 味药材进行鉴别。
4.3 含量测定指标选择 预实验发现,君药苍术指标成分苍术素易挥发,在提取、样品处理过程中含量太低而检测不到;白术内酯类成分是白术发挥降糖作用的有效成分,但其化学性质不稳定[19-21],容易发生转化,并且对照品价格较高。因此,本实验选择臣药及其他药材有效成分进行测定。
4.4 流动相选择 本实验参考文献[22-24] 并经反复摸索,确定甲醇-0.1%磷酸作为流动相,并且采用梯度洗脱,此时各成分分离度均大于1.5,理论塔板数均符合要求。
4.5 检测波长选择 各成分吸收波长分布在238、250、327 nm 处,其中238 nm 处吸收峰峰值较高,但出峰数量较少;327 nm 处色谱峰峰值较低;250 nm 处色谱峰数量较少。经反复摸索,最终选择294 nm 作为中间值,在保证色谱峰数量的同时峰值也不会太低。
5 结论
本实验所建立的消渴健脾胶囊制备工艺符合要求,可投入生产,并且其质量标准合理可靠,可为后续相关研究提供一定参考。
