高效液相色谱法检测微拟球藻中磷脂类化合物含量
2023-09-18刘耘珲周真真张俊杰
刘耘珲,周真真,陈 林,张俊杰,丛 威
(1.江苏海洋大学,江苏 连云港 222000;2.中国科学院过程工程研究所,北京 100089;3.中国科学院青岛生物能源与过程研究所,山东 青岛 266101)
微拟球藻(Nannochloropsis sp.)是一种高附加值光合藻类微生物,能够通过光合作用合成脂质、蛋白质、叶绿素和类胡萝卜素等高价值产物[1]。该藻种总脂含量可达干重的30%~50%[2-5],其中磷脂的含量可达30%[6],同时还富含大量的ω-3 系列的多不饱和脂肪酸,尤其是二十碳五烯酸(eicosapentaenoic acid,EPA),具有预防心血管疾病等功能[7-9],相比传统油料作物中提取的磷脂,微拟球藻所提取的磷脂具有更高的营养价值。因此,深入研究微拟球藻中磷脂的组成,对于后期磷脂产品的开发和利用具有重要意义。
目前,磷脂检测主要采用高效液相色谱法,这种方法具有便捷高效且准确的优点[10-17]。由于磷脂是一种亲水性物质,因此使用HILIC 反相分离模式的ZORBAX RX-SIL 色谱柱能够有效地分离微拟球藻磷脂的各个组分。微拟球藻磷脂含有长链、高度不饱和的脂肪酸基团,并且含有较高的细胞色素、蛋白质和多糖等杂质。采用大豆或鸡蛋等传统的磷脂检测方法可能会影响检测的准确性。因此,针对微拟球藻磷脂体系,建立高效液相色谱分析方法具有重要意义。
本研究使用ZORBAX RX-SIL 色谱柱和紫外检测器对磷脂组分进行分离检测,考察了不同的流动相对磷脂的检测效果,以期建立高效且准确的微拟球藻磷脂检测方法,对微藻源磷脂的检测提供参考。
1 材料与方法
1.1 材料与试剂
微拟球藻(山东烟台海融生物技术有限公司);乙腈(色谱纯,美国Sigma 公司);甲醇(色谱纯,美国Sigma 公司);磷脂酰肌醇(PI,批号H2219476,纯度大于99%,上海阿拉丁生化科技股份有限公司);磷脂酰乙醇胺(PE,批号E2113225,纯度大于99%,上海阿拉丁生化科技股份有限公司);磷脂酰胆碱(PC,批号D2101126,纯度大于98%,上海阿拉丁生化科技股份有限公司);溶血磷脂酰胆碱(LPC,纯度大于99%,批次C2212077,上海阿拉丁生化科技股份有限公司);硅胶固相萃取柱(Waters Sep-PakTM 6cc 500 mg)。
1.2 仪器与设备
Agilent 1200 Ⅱ高效液相色谱仪带紫外检测器(美国安捷伦公司);FA2004 电子分析天平(上海精科仪器有限公司);NK200-1B 氮吹仪(杭州米欧仪器有限公司);旋转蒸发仪;TGL-16C 冷冻离心机(上海安亭科学仪器厂)。
1.3 实验方法
1.3.1 标准储备液的配制
分别精密称取磷脂酰肌醇、磷脂酰乙醇胺、磷脂酰胆碱和溶血磷脂酰胆碱标准品25 mg 置于容量瓶中,用甲醇溶解并定容至25 mL,此时配制的标准储备液中磷脂酰肌醇、磷脂酰乙醇胺、磷脂酰胆碱和溶血磷脂酰胆碱的浓度均为1 mg/mL。分别取上述4 种标准品1 mL 配制成混合标准溶液。
1.3.2 标准工作曲线溶液的配制
精密吸取上述磷脂酰肌醇、磷脂酰乙醇胺、磷脂酰胆碱和溶血磷脂酰胆碱标准储备液,用甲醇稀释,分别配制浓度为0.1、0.2、0.4、0.6、0.8、1 mg/mL 的标准溶液。
1.3.3 微拟球藻磷脂样品的制备
微拟球藻藻粉经膨化后得到微拟球藻膨化颗粒,首先取100 g 膨化后的藻粉,加入300 mL 95%乙醇,放置于磁力搅拌器上,在60 ℃的条件下500 r/min 提取1 h,然后离心样品收集上清液;再向膨化藻粉沉淀中加入100 mL 95%乙醇,重复上述提取过程2 次,合并上清液。最后使用0.22 μm 有机系过滤器过滤,将滤液置于旋转蒸发仪中去除溶剂,得到微拟球藻油脂。
称取约50 mg 上述油脂样品,溶于1 mL 氯仿准备上样。取硅胶小柱,先用10 mL 甲醇活化,再加入30 mL 氯仿平衡,然后将油脂样品上样。经过25 mL 氯仿、20 mL 丙酮和15 mL 甲醇先后洗脱,分别获得中性脂、糖脂和磷脂组分,收集磷脂组分,利用氮吹仪除去磷脂组分中的溶剂,并烘干至恒重[18]。最后精密称取10 mg微拟球藻磷脂,用甲醇溶解并定容至10 mL 容量瓶中,经0.45 μm 微孔滤膜过滤,置于冰箱-20 ℃保存备用。
1.3.4 仪器参考条件
色谱柱为ZORBAX RX-SIL 柱(4.6 mm×250 mm,5 μm);检测波长205 nm;流动相为甲醇、甲醇-乙腈溶液(50∶50,V∶V)和甲醇-水溶液(87∶13,V∶V),等度洗脱;流速为1 mL/min;柱温为30 ℃;进样量为10 μL。
1.3.5 精密度实验
取4 种磷脂混合标品重复进样3 次,分别测得峰面积并计算RSD 值,检测仪器的精密度。
1.3.6 加标回收率实验
取已制备好的已知浓度的微拟球藻磷脂样品溶液,向其中加入已知质量的磷脂标准品,制成低、中、高3 种浓度加标回收样品,分别进样平行测定3 次,计算磷脂的加标回收率。
1.3.7 数据分析
采用色谱处理软件,根据色谱图中样品的保留时间和标准品的保留时间比对进行定性分析,根据各磷脂的峰面积和标准曲线对样品中的磷脂进行定量分析。用Microsoft Excel 2016、Origin 9.8 软件对实验数据进行处理。
2 结果与分析
2.1 色谱条件的选择
2.1.1 流动相的选择
考察甲醇、甲醇-乙腈溶液、甲醇-水溶液作为流动相对微拟球藻磷脂样品中4 种目标物质的影响。由图1可知,当采用甲醇-乙腈溶液(50∶50,V∶V)作为流动相时,2~3 min 处的峰无法较好地分离,且目标物的保留时间加长,峰形较差;当采用甲醇作为流动相时,2~3 min处的峰无法较好地分离,并且检测时间较长,需要15 min;当采用甲醇-水溶液(90∶10,V∶V)作为流动相时,基线平稳,目标峰形较好,且检测时间缩短至8 min 以内。因此,选用甲醇-水溶液作为流动相进行后续的实验。
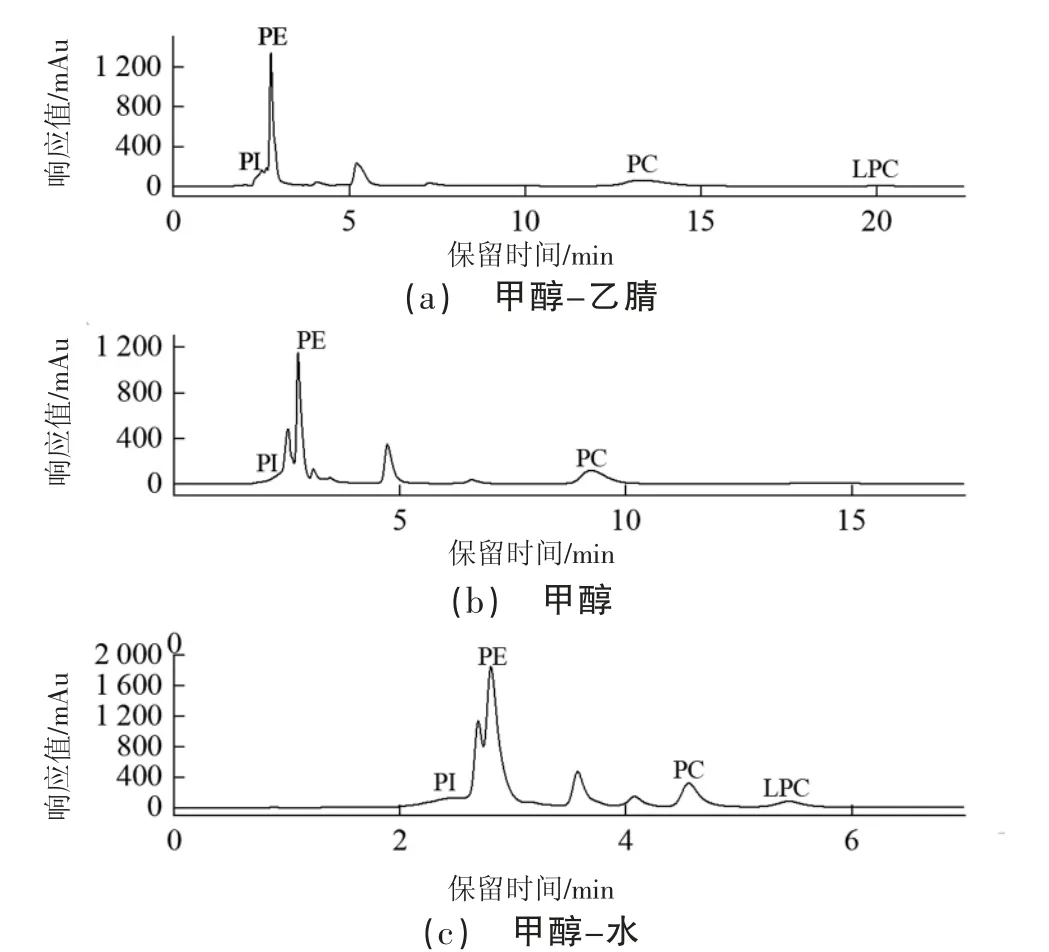
图1 微拟球藻磷脂样品在不同流动相下的液相色谱图
2.1.2 流动相比例的选择
考察流动相为甲醇-水溶液时水的比例分别为5%、10%、13%、15%、20%时对微拟球藻磷脂样品中4种目标物质的影响,结果如图2 所示。随着水的比例增加,样品中目标物的保留时间缩短。当水的比例较低时,2 min 处的PI 无法有效分离;当水的比例过高时目标物的峰会发生重叠。因此,选择水的比例为13%进行后续实验。

图2 微拟球藻磷脂样品在不同甲醇-水比例下的液相色谱图
2.2 方法学验证
2.2.1 线性关系、检出限与定量限
精密吸取标准工作曲线溶液进样测定,以各个标准品质量浓度(X)为横坐标,对应的峰面积(Y)为纵坐标绘制标准曲线,回归方程见表1,4 种组分在0.1~1 mg/mL 的范围内线性关系良好,相关系数大于0.996 8;分析方法的定量限和检出限由信噪比(S/N)计算,检出限定义为S/N=3 时对应的分析物浓度,定量限定义为S/N=10 时对应的分析物浓度,分别计算4 种组分的检出限和定量限。
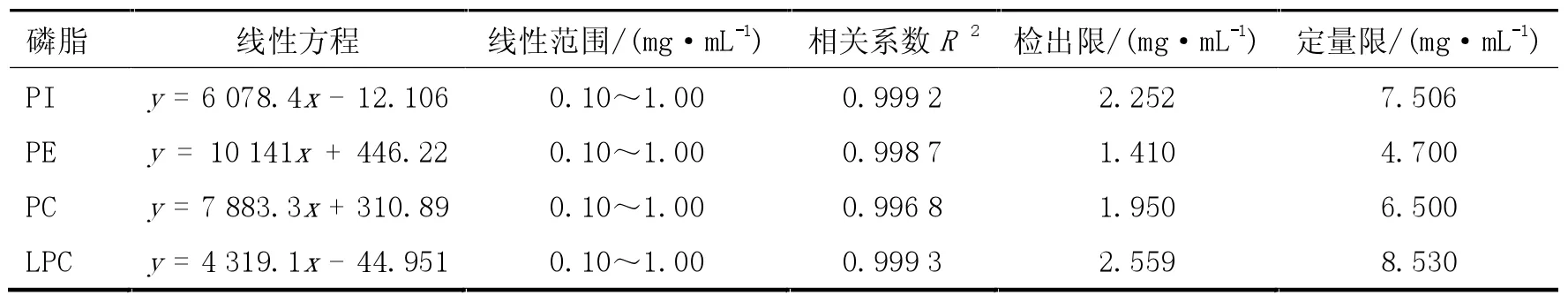
表1 各磷脂组分的线性方程、范围、相关系数、检出限及定量限
2.2.2 精密度
精密度结果见表2,结果显示4 种磷脂标准品的相对标准偏差(RSD)均小于0.48%,重现性良好。
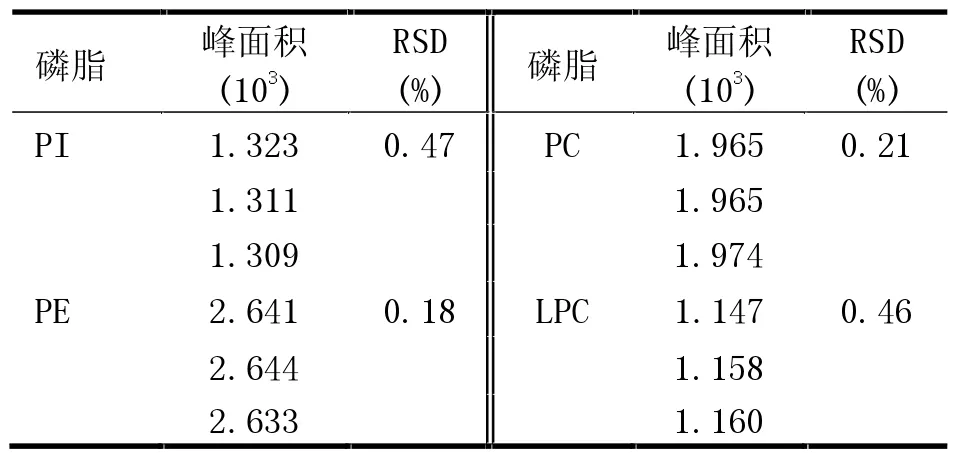
表2 磷脂标准品精密度实验结果
2.2.3 回收率
回收率结果见表3,在不同添加水平下,4 种磷脂标准品的回收率在85%~105%,相对标准偏差(RSD)小于3.1%,有较好的准确度。

表3 磷脂标准品加标回收率实验结果
2.3 样品检测
按照1.3 中的方法对微拟球藻磷脂样品进行检测,检测峰面积代入线性回归方程中计算磷脂酰肌醇、磷脂酰乙醇胺、磷脂酰胆碱和溶血磷脂酰胆碱的含量,结果见表4。计算结果为磷脂酰肌醇含量为19.32%,磷脂酰乙醇胺含量为18.74%,磷脂酰胆碱含量为11.86%,溶血磷脂酰胆碱含量为3.24%。
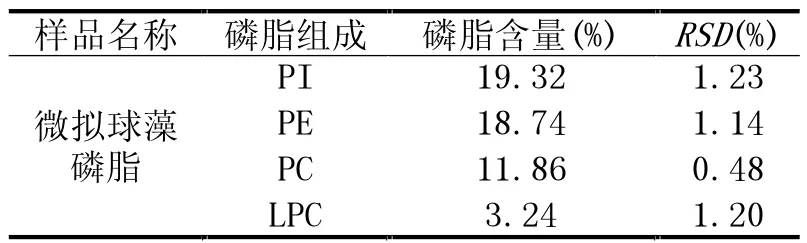
表4 微拟球藻磷脂中PI、PE、PC 和LPC 含量
3 结论
在研究制定高效液相色谱方法时,流动相的选择是影响分离效果的关键因素之一[19]。为了分析微拟球藻磷脂类化合物的组成,本研究建立了一种以甲醇-水(87∶13,V∶V)为流动相,采用ZORBAX RX-SIL 色谱柱和紫外检测器的反相高效液相色谱法。结果表明,微拟球藻中最主要的4 种磷脂类化合物磷脂酰肌醇、磷脂酰乙醇胺、磷脂酰胆碱和溶血磷脂酰胆碱含量分别为19.32%、18.74%、11.86%和3.24%。经过方法学验证,该检测方法的加标回收率在85%~105%之间,灵敏度较高,检测时间较短,分离度较好,且具有良好的稳定性,适用于微藻磷脂的测定。
