Vohwinkel综合症分子遗传学研究进展
2023-09-16王震英
凌 霞 王震英 张 莉
1.山东第一医科大学附属山东省立医院皮肤科,山东 济南 250021;2.山东第一医科大学(山东省医学科学院)研究生部,山东 济南 250117
Vohwinkel 综合症(Vohwinkel syndrome,VS)又名残毁性掌跖角皮病(mutilanting palmoplantar keratoderma)、残毁性遗传角质瘤(keratoma hereditaria mutilans),沃温克综合征。最早由德国学者Vohwinkel 于1929 年描述并命名,该病为罕见遗传性皮肤病,是掌跖角化病的一种,多为常染色体显性遗传病,也有隐性遗传的报道。典型临床表现包括弥漫性掌跖角化过度,蜂巢样外观,海星状或条状角化过度斑块,指(趾)远端呈环形缩窄并逐渐断离[1],严重者出现肢端残缺。该病治疗困难,多为对症治疗,可以系统应用类维甲酸如阿维a 和异维甲酸等来缓解角化过度[2-4],但是出现收缩带和自动截肢时则需要切除收缩环,移植或皮瓣移植等手术治疗[2,5]。本病临床表现复杂,常伴有听力损失或鱼鳞病,伴有听力损失者被称为典型VS;伴有鱼鳞病者被称为变异型VS(OMIM 604117)。也有同时伴有先天性耳聋和鱼鳞病的病例[6]。VS 组织病理表现为:角化亢进伴角化不全,颗粒层增厚,棘层肥厚,真皮浅层炎细胞浸润[1]。VS的发生多与角质形成细胞的异常增殖和凋亡相关,可能与AP-1,STAT等分子的异常有关,但具体涉及的分子及机制,知之甚少。目前多为关于VS的致病基因和诊疗方面的综述[7],缺少与该病相关的基因功能学的描述。本文着重于每个突变体及其功能的改变,试图从这些突变中总结这些基因在维持正常皮肤功能方面的可能作用,为其治疗提供新的思路。
近年来诸多学者对该病致病基因进行研究,发现2个VS的相关位点,1个是位于染色体13q11-q12的GJB2(gap junction protein beta 2),编码连接蛋白26(connexin 26,Cx26)。另1 个是位于染色体1q21的LOR(loricrin),编码兜甲蛋白。GJB2突变常伴听力损害,导致经典型VS,而LOR突变常伴有鱼鳞病,导致变异性VS。目前发现的与VS 有关的GJB2突变如下:c.196G > C (p.Asp66His)、c.175G > a (p.Gly59Ser)、c.193T > C(p.Tyr65His)、c.389G > T(p.Gly130Val)和c. 358-360delGAG(delE120)[8],多为错义突变(表1)。与VS 有关的LOR 突变如下:730insG、662insT、709insC 和c. A796G(p. Ser266 Gly)(表2)。

表1 Vohwinkel综合症GJB2 5种突变体对比

表2 Vohwinkel综合症LOR 4种突变体对比
1 GJB2
GJB2全长4 804 bp,编码Cx26。Cx26包含跨膜区(M1-M4)、细胞膜外区(E1,E2)、胞浆内连接区(CL)、细胞质氨基末端(N 末端)和羧基末端(C 末端)。N端区和跨膜区M1的细胞内侧端构成充当电压感受器的电荷复合体,细胞外区E1 和E2 决定缝隙连接通道与其他连接蛋白的相容性,胞内连接区和C 端区与缝隙连接通道pH 值的门控有关。6 个连接蛋白寡聚化形成中空六聚体,称为连接子或半通道,然后转位到细胞质膜。位于相邻2 个细胞膜上的2 个连接子通过2 个胞外域对接,组成亲水的低电阻通道[9]。这些通道是细胞间电解质、第二信使和代谢物质的重要通路,在信息传递和物质交换中起重要作用。连接蛋白在内耳和表皮中表达,在维持内耳钾稳态和表皮角质形成细胞的生长、分化中起重要作用[1]。皮肤表达的缝隙连接蛋白基因突变会破坏表皮的生长和分化(干扰表皮分化和过度角化)。Bakirtzis 等[10]通过转基因技术构建了GJB2p.Asp66His突变过表达的小鼠模型,该模型模拟了真实的VS。突变小鼠表皮连接蛋白丢失,在细胞质中积累,表皮角化层明显增厚。该模型研究表明,VS(D66H)突变型病理机制可能涉及组织缝隙连接网络的直接破坏,也可能涉及突变蛋白之间的新型相互作用,导致细胞死亡。
目前与VS 有关的GJB2突变体有5 种,其中GJB2的c. 196G > C (p. Asp66His)突变[11]、c. 175G > A (p. Gly59Ser)突变[12]和c. 193T > C(p.Tyr65His)[13]突变均发生在E1 区(图1A,1B),影响了通道通透性,钙等物质转移减少。其中GJB2的p. Asp66His 突变编码的Cx26 不产生通道功能,与连接蛋白43 共定位,相互作用[14]。Cx26 局部缺失或连接蛋白30缺失只会产生耳聋,不会产生皮肤病表型,说明连接蛋白的功能存在冗余。经典VS 皮肤表型的产生可能是由于突变基因编码的蛋白的运输缺陷和通道功能的改变引起的。GJB2的c.389G > T(p.Gly130Val)突变位于细胞内的第2个结构域[15],与以往突变不同,轻度PPK 和严重耳聋之间存在表型差异,差异的原因仍不清楚,或可从突变基因功能改变的研究找到合理的解释。GJB2的c. 358-360delGAG(delE120)突变为纯合子突变[16]。
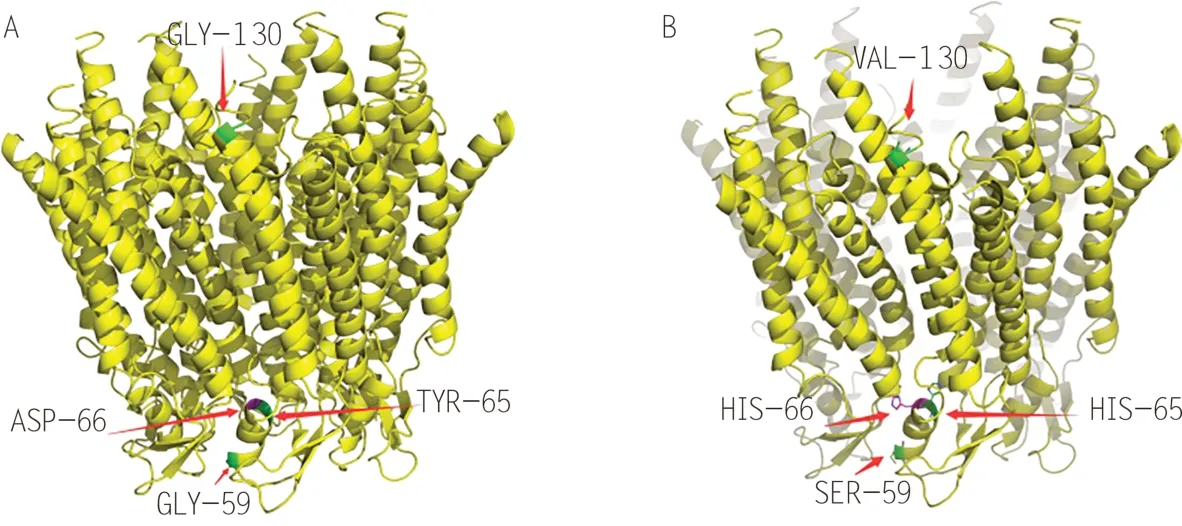
图1 Cx26缝隙连接通道结构及其突变位点(其中120位点Glu缺失突变在结构信息里面缺失)
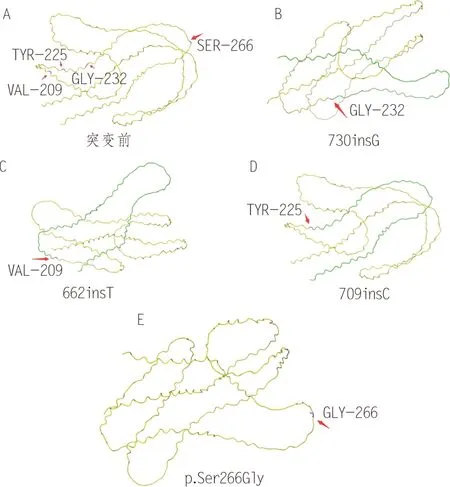
图2 兜甲蛋白结构及其突变位点
引起经典VS 的GJB2突变具有共同的分子表型,它们大多(除了p.Gly130Val)聚集在Cx26 的E1结构域,影响蛋白运输和通道通透性。其中芳香烃受体(aryl hydrocarbon receptor, AHR)信号通路、干扰素(interferon, IFN)信号通路、丝裂原活化蛋白激酶(mitogen activated protein kinase, MAPK)信号通路的分子的异常可能参与不同GJB2突变型所引起的VS 的发病[8]。此外,VS 中蛋白酪氨酸激酶/信号传导及转录激活蛋白(janus kinase-signal transducers and activators oftranscription, JAK/STAT)通路被预测为强烈激活[8]。JAK/STAT 是一种普遍表达的细胞内信号转导通路,涉及包括细胞増殖、分化、凋亡和免疫调节在内的许多关键的生物学过程[17]。该通路在系统性红斑狼疮、特应性皮炎、斑秃、扁平苔藓、皮肌炎、移植物抗宿主病、化脓性汗腺炎、银屑病和白癜风[18]等与炎症和免疫相关的皮肤疾病的发病机制和治疗领域研究的较为深入。通过抑制JAK 信号通路可抑制银屑病中细胞的增殖和角化,调节炎症反应[19-20]。VS中JAK1和STAT2的mRNA 表达量上调,激活JAK/STAT 信号通路可能在调控VS的角质形成细胞的异常增殖和角质化中起作用[8],但其在GJB2突变引起的VS 皮肤症状中具体扮演的角色,以及与其他调控子或信号因子的更为具体的相互作用关系,有待进一步研究。
此外,Decrock 等[21]认为,细胞凋亡不仅与缝隙连接相关的信号通路改变有关,也可能通过缝隙连接蛋白介导的细胞凋亡前后抗凋亡分子的扩散来控制细胞的存亡。Zhang 等[22]发现,GJB2基因敲除小鼠耳蜗外毛细胞表现了典型的凋亡态。GJB2基因敲除小鼠耳蜗感觉上皮细胞发生凋亡[23]。
2 LOR
LOR位于染色体1q21的表皮分化复合物区域,编码兜甲蛋白,该基因突变会导致变异型VS。角质化包膜(cornified envelope, CE)中的成分以复杂的、多阶段相互作用沉积在质膜的内表面是形成正常角化表皮的关键。兜甲蛋白是CE 中的一种蛋白,能维持皮肤角质层的完整性和皮肤屏障的稳定性。兜甲蛋白由4 个甘氨酸环结构域组成,这些结构域的两侧是富含谷氨酰胺和谷氨酰胺/赖氨酸的区域。甘氨酸环结构域是高度灵活的,“velcro”假说认为,角蛋白的甘氨酸环与兜甲蛋白的甘氨酸环通过弱疏水性和氢键相互作用结合在一起,当施加适当的压力时就会破坏这种相互作用,并诱导他们之间产生一种新的类似的相互作用,当除去压力时,这种新的相互作用又会消失[24]。该作用一定程度解释了表皮的柔韧性和弹性恢复特征,可能赋予CE 和表皮灵活性和延展性;而谷氨酰胺和谷氨酰胺/赖氨酸区域是谷氨酰胺转胺酶介导的Nε-(γ-谷氨酰胺基)赖氨酸异二肽交联CE组分的关键底物,这是CE高度不溶性的原因。变异的兜甲蛋白包含一个以基本氨基酸短链延伸为特征的羧基末端序列,结构扭曲,无法产生明显二级结构,破坏交联,无法进入角质层,但是会进入颗粒层细胞核内并逐渐积累,会导致表皮间水分的流失和鱼鳞病[8],干扰角质化细胞终末分化。
LOR第730 位插入核苷酸G 致密码子232 框移的杂合突变是最常见的一种突变,蛋白异常易位到细胞核内[25]。LOR的662insT杂合突变[26],第4个甘氨酸环结构域和富含谷氨酰胺/赖氨酸的c 端结构域被一个富含精氨酸和亮氨酸的区域取代。这2种突变都破坏了甘氨酸环,可能通过破坏谷氨酰胺转胺酶介导的其与自身或其他CE成分的交联能力或者通过使其结构僵化,削弱其灵活性而破坏包膜的作用;突变也可能导致C端肽中富含精氨酸的序列中引入潜在的核靶向基序,这种序列的存在或可解释颗粒细胞层中兜甲蛋白在核内积累[27-28]。兜甲蛋白单体之间可以通过二硫键相互交联,VS患者核内除了异常兜甲蛋白,还有正常的兜甲蛋白,2种兜甲蛋白之间可以交联,在核内积累,会导致相对的兜甲蛋白缺乏,并进一步导致有缺陷的CE 形成[29]。Ishida-Yamamoto 等[29]在一个日本VS 患者中发现了LOR第709 位后面插入了一个C 残基,突变导致移码使C端的91个氨基酸变成了错义氨基酸,错义氨基酸清除了约1/3参与异二肽交联形成的谷氨酰胺和赖氨酸残基;兜甲蛋白和其他角化细胞包膜蛋白之间的分子间交联的相对数量被突变破坏。LOR存在p.Ser266Gly 突变的患者既有先天性感音性耳聋[6],也伴有鱼鳞病。该突变在既往插入突变高发区的肽链的C 末端,该区可能是LOR重要的功能区,突变使丝氨酸转变为甘氨酸,甘氨酸较丝氨酸在结构上少一个醛基结构,突变可能影响到蛋白质的空间构象,从而引起功能的改变。但丝氨酸和甘氨酸性质基本相同,而且引起耳聋的原因尚未明确,可能还存在其他的致病基因,需要进一步进行功能方面的实验深入探讨。
LOR突变多为插入突变,位于LOR的C 端结构域,影响甘氨酸环,破坏兜甲蛋白的细胞定位和交联作用。Yoneda 等[30]发现,表皮生长因子受体(epithelial growth factor receptor, VEGF)、血管内皮生长因子受体2(vascular endothelial growth factor receptor 2, VEGFR2)和信号传导与转录激活因子3(signal transducer and activator of transcription 3,STAT3)在LOR突变的细胞中均发生磷酸化,STAT3蛋白在突变体细胞中与VEGF 启动子结合,提示在loricrin 角化病细胞模型中,VEGF 的释放和随后VEGFR2的激活将LOR突变与细胞的快速增殖联系起来。这些分子在上述GJB2突变导致的VS中也出现了异常,可能是不同VS的共同致病通路。
3 总结与展望
VS具有临床和遗传异质性,不仅编码不同的基因突变表型存在差异,编码相同的GJB2突变产生的表型也存在差异,如p.Gly59Ser突变体引起的VS可同时伴有鱼鳞病和遗传性耳聋,而p.Asp66His突变体引起的VS仅伴有耳聋,差异的原因尚不清楚。经典VS和变异型VS都存在致病基因编码蛋白质的错误定位[30],可能会引起缝隙连接的形成,进而影响信号分子的传导。值得注意的是,郭可盈等[8]通过基因芯片发现了经典VS 中JNK/AP-1 通路的异常。AP-1 因子参与调控角质形成细胞的增殖、分化、凋亡和转化,AP-1 信号分子的改变可能是多种类型角化病的共同通路。在无LOR遗传背景下,抑制AP-1(c-Jun)功能的小鼠会出现变异型VS 表型[31];小鼠表皮AP-1(c-Jun 和JunB)的抑制导致银屑病样异常,包括关节炎症,角化过度、角化不全与T细胞和中性粒细胞浸润[32];Tang等[33]发现,在过表达连接蛋白31导致的角质形成细胞过度增生、角化不全、角化过度、淋巴细胞浸润的家族性变异型红皮角化病中c-Fos/JunB 表达增加,AP-1 抑制剂的使用使表型得到了改善。但是该信号通路在VS中的作用知之甚少,具体机制需进一步研究。对于变异型VS,LOR的多态性或者存在不同的修饰基因,影响了VS 的发病机制和表型。Yoneda 等[30]发现的VEGF、VEGFR2和STAT3分子的异常与角质形成细胞异常增殖可为研究VS的致病机制提供思路。病情严重时可致残致畸,危害患者生活,当下基因治疗还处于起步阶段,基础研究与临床使用还有很大的距离,分子遗传学研究疾病的发病机制将为其治疗提供新的思路,随着研究的深入,相信将来会使患者获得更大的受益。
利益冲突所有作者均声明不存在利益冲突
