多重耐药基因lsa(E)实时荧光定量PCR检测方法的创新
2023-09-04张博陈峥李琼刘伟成杜聪阳杜向党许春燕
张博,陈峥,李琼,刘伟成,杜聪阳,杜向党,许春燕
(河南农业大学动物医学院,河南 郑州 450046)
细菌耐药已成为全球公共卫生问题,严重威胁食品安全和公共健康[1]。多重耐药基因lsa(E)属于能够编码ATP结合盒(ATP-binding cassette,ABC)转运蛋白的基因,通过外排作用介导革兰氏阳性菌感染的代表药物(林可胺类、链阳菌素A类和截短侧耳素类)的抗性[2]。此外,lsa(E)基因还可介导阳性菌株对人类医学临床上重要的抗革兰氏阳性菌药物来法莫林(lefamulin)[3]和瑞他莫林(retapamulin)[4]的耐药性。多重耐药基因lsa(E)由德国STEFAN Schwarz教授于2012年首次在欧洲人源耐甲氧西林金黄色葡萄球菌(methicillin-resistant staphylococcus aureus,MRSA)和甲氧西林敏感的金黄色葡萄球菌(methicillin-resistant staphylococcus aureus,MSSA)中被检出并命名[2]。同年,LI等[4]在江苏和浙江猪场MRSA中检测到lsa(E)基因,并证实该基因位于多重耐药基因簇中。随后,该基因又在多种细菌中被发现,包括粪肠球菌[5]、屎肠球菌[5]、表皮葡萄球菌[6]、无乳链球菌[7]和猪红斑丹毒丝菌[8]等。目前,lsa(E)基因相继在世界各地被报道,包括亚洲、欧洲、南美洲和北美洲等[2,7,9-12]。此外,lsa(E)基因可以通过质粒在不同细菌之间传播[4-6],这加速了其散播的速率和广度,对畜禽养殖业和人类健康带来威胁。
在耐药基因的相关研究中,普通聚合酶链式反应(polymerase chain reaction,PCR)检测方法或多重PCR检测方法只能对其进行定性检测,检测对象限定为纯培养菌株的基因组,并在检测之后还需要进行测序验证。谷立慧等[13]使用普通PCR方法与测序技术相结合对猪源金黄色葡萄球菌lsa(E)基因的流行情况进行调查,需要先使用PCR对纯培养物进行扩增,然后对扩增产物进行测序验证。随着耐药基因和耐药菌株作为新型污染物概念的出现,实时荧光定量PCR(real-time quantitative PCR,qRT-PCR)和宏基因组学等非培养的检测方法已被用于表征各种环境介质中耐药基因的污染水平。然而,宏基因组成本高,仍未被广泛应用。而荧光定量PCR敏感性高[14],特异性和重复性好[15],成本低廉,可以被广泛应用于微生物诊断检测的研究中。目前,常见的检测方法并不能满足畜禽养殖环境中lsa(E)基因定量检测的需求,而关于lsa(E)基因qRT-PCR检测方法尚未报道。基于此,本研究创新了lsa(E)基因的qRT-PCR检测方法,并使用该方法检测牛养殖环境中lsa(E)基因的拷贝数量和相对丰度,为畜禽养殖环境中lsa(E)基因的快速精准检测提供了理论依据。
1 材料与方法
1.1 试验材料
1.1.1 材料 河南农业大学病原菌耐药与新兽药创制实验室分离保存的1株携带lsa(E)基因的粪肠球菌(Enterococcusfaecalis)a90为阳性对照菌株,以其基因组作为阳性对照模板。2021年9月在河南省中牟县2个牛养殖场采集牛场粪便(NNF1~NNF3,RCF1~RCF3,NBF1~NBF3,RDF1~RDF3)、卧料(NNW1~NNW3,NBW1~NBW3)、土壤(RDT1~RDT3)和淤泥(RDY1~RDY3)共24份环境样品。
1.1.2 主要试剂 细菌基因组DNA提取试剂盒,天根生化科技有限公司;HiPure Soil DNA Mini Kit,广州美基生物科技有限公司;QIAGEN ©Plasmid Mini Kit,德国QIAGEN公司;PowerUPTMSYBRTMGreen Master Mix(2×),北京赛默飞世尔科技(中国)有限公司;胶回收纯化试剂盒,北京天根生化科技有限公司;One step ZTOPO-Blunt/TA零背景快速克隆试剂盒,北京庄盟国际生物基因科技有限公司;DH5α感受态细胞,北京天根生化科技有限公司。
1.1.3 主要仪器 qRT-PCR仪(CFX96),美国Bio-Rad公司;PCR仪(T100 Thermal Cycler),美国Bio-Rad公司;凝胶成像系统(Tanon 1600),上海天能科技有限公司;电泳仪(HE-120),上海天能科技有限公司;超微量分光光度计(NanoDrop ND-2000),美国Thermo公司;荧光定量仪(Qubit 4.0),美国Thermo公司。
1.2 试验方法
1.2.1 引物的设计与合成 通过NCBI查找并下载lsa(E)基因的参考序列,用软件Oligo7进行qRT-PCR特异性引物设计,lsa(E)基因qRT-PCR引物(基因序列号67412470)F:ACCAAAATAAACCCCTTCTTGCGTT,R:CCTGCTTATTGATGAACCTACAAACCA,片段长度为93 bp;T载体引物(M13引物),F:TGTAAAACGACGGCCAGT,R:CAGGAAACAGCTATGACC,片段长度为237 bp;lsa(E)基因普通PCR引物[16]:F:TGTCAAATGGTGAGCAAACG,R:TGTAAAACGGCTTCCTGATG,片段长度为496 bp;16S rRNA基因引物参考已发表文献[17]进行合成,F:CGGTGAATACGTTCTCGG,R:GGATACCTTGTTACGACTT,片段长度为143 bp。
1.2.2 阳性标准质粒的制备 将保存的lsa(E)基因阳性肠球菌a90扩大培养,参照DNA提取试剂盒说明书提取DNA,以其为模板进行PCR扩增lsa(E)基因,反应体系为:模板2 μL,上、下游引物各1 μL,Premix Ex taq DNA聚合酶10 μL,ddH2O 6 μL,总体积为20 μL。反应程序为:94 ℃预变性5 min;94 ℃ 30 s,58 ℃ 30 s,72 ℃ 15 s,进行30个循环;72 ℃延伸10 min;4 ℃保存备用。
通过常规方法,将目的片段lsa(E)基因与T载体连接,构建重组质粒。测序验证之后将重组质粒电转化入大肠杆菌DH5α中,提取质粒后使用Qubit 4.0荧光定量仪检测质粒DNA质量浓度,NanoDrop ND-2000超微量分光光度计确定质粒DNA纯度,-20 ℃保存备用。
1.2.3lsa(E)基因qRT-PCR方法设计 使用Qubit 4.0对重组质粒定量后,根据下式计算质粒拷贝数量[18]。将重组质粒质量浓度稀释至30~40 mg·L-1,之后进行10倍的倍比稀释,获得重组质粒的10个稀释点,即10-1、10-2、10-3、10-4、10-5、10-6、10-7、10-8、10-9、10-10;每个点设置3个平行[19],另设超纯水(ddH2O)组作为阴性对照,按照式(1)计算拷贝数量。
CN=(L×C)/(N×M×109)
(1)
式中:CN为拷贝数量/(Copies·μL-1);L为阿伏伽德罗常数/(6.02×1023mol-1);C为重组质粒质量浓度,36.8 mg·L-1;N为重组质粒的总长度,1 958 bp;M为每对碱基的平均摩尔质量,660 g·mol-1。
反应体系为:SYBRTMGreen Master Mix(2×)10 μL,lsa(E)基因的上、下游引物各0.8 μL,ddH2O 6.4 μL,DNA模板2 μL。反应程序如下:(50 ℃,2 min)+(95 ℃,3 min)+(95 ℃,30 s+58 ℃,30 s+72 ℃,10 s)×40;熔解曲线的反应条件是(95 ℃,30 s)+(60 ℃,30 s)+(72 ℃,30 s)。反应后整理相关数据,以标准品拷贝数量的对数值为横坐标,qRT-PCR得到的Ct值为纵坐标,绘制lsa(E)基因的标准曲线。
1.2.4 qRT-PCR方法的重复性试验 qRT-PCR反应选择同一批3个不同质量浓度的标准品进行批内重复试验;用不同批次的3个质量浓度的标准品为模板,在不同时间内进行批间重复试验,运用变异系数数值,即标准差与平均值的比值,计算批内批间变异系数。
1.2.5 牛场环境样品中lsa(E)基因拷贝数量和相对丰度的检测 使用HiPure Soil DNA Mini Kit试剂盒对24份环境样品的宏基因组进行提取。以提取的宏基因组为模板,使用1.2.1中lsa(E)基因和16S rRNA基因的qRT-PCR引物,按照1.2.3的反应体系和反应程序检测lsa(E)基因和16S rRNA拷贝数量,并计算lsa(E)基因的相对丰度。相对丰度=lsa(E)基因拷贝数量/16S rRNA拷贝数量。
1.2.6 普通PCR对牛场环境样品中lsa(E)基因的检测 以1.2.5中提取牛场环境样品的宏基因组为模板,使用1.2.1中lsa(E)基因普通PCR引物,对1.2.5中24份环境样本中lsa(E)基因进行检测。
1.2.7 数据统计与分析 试验数据采用Excel 2019进行数据处理和相关分析,用平均值±标准差表示。
2 结果与分析
2.1 lsa(E)基因的PCR扩增
使用1.2.1中lsa(E)基因引物进行PCR扩增。如图1所示,阳性孔中只有1条明亮的条带,无杂带,条带大小与设计大小一致,为93 bp,阴性对照未出现条带,说明本试验设计的引物特异性良好,无引物二聚体出现。
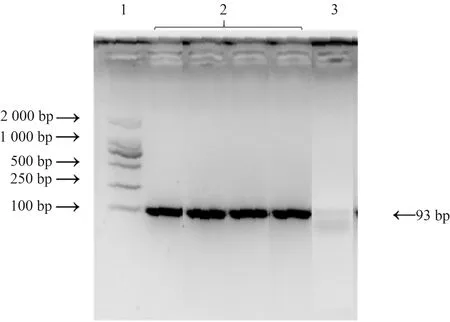
1.2 000 bp DNA maker;2.lsa(E)基因片段;3.阴性对照。
2.2 lsa(E)基因的克隆和重组质粒的制备
利用1.2.2中lsa(E)基因引物和M13引物对挑取的转化子进行PCR验证,确定重组质粒是否构建和转化成功。结果如图2所示,在电转化子中,lsa(E)基因扩增的片段长度为93 bp,引物M13扩增的片段长度为237 bp,说明lsa(E)基因片段与T载体连接成功并转化入大肠杆菌DH5α。利用紫外分光光度计检测该重组质粒的质量浓度,结果为36.8 mg·L-1。该质粒作为阳性标准品,根据1.2.3中的公式计算其拷贝数量,计算结果为1.71×1010copies·μL-1。

1.2 000 bp DNA maker;2.lsa(E)基因片段;3.质粒引物M13扩增验证;4.阴性对照。
2.3 lsa(E)基因qRT-PCR标准曲线,扩增曲线和熔解曲线结果
将构建的重组质粒以10倍梯度稀释之后,进行qRT-PCR,并扩增lsa(E)基因,得到标准曲线(图3-A)、扩增曲线(图3-B)和熔解曲线(图3-C)。综合性分析可知,标准曲线的拷贝数量和Ct值呈现线性反比关系。根据数据计算lsa(E)基因回归方程,结果为y=-3.285 2x+42.137,且R2为0.997,表明标准品在此范围内线性关系好,lsa(E)基因的检测拷贝数量范围是1.71×102~1.71×108copies·μL-1,Ct值的范围是15.22~34.27。lsa(E)基因标准曲线的扩增效率为101.56%,处于90%~110%,符合要求。扩增曲线平滑,不同质量浓度标准品的扩增曲线平行分布,熔解曲线呈单峰,表明本试验设计的引物特异性强,无非特异性扩增和引物二聚体的存在。因此,该检测方法良好,能够用于后续环境样品中lsa(E)基因拷贝数量的检测。

A.标准曲线;B.扩增曲线;C.熔解曲线。
2.4 qRT-PCR方法的重复性试验测试结果
以质粒拷贝数量1.71×103~1.71×105copies·μL-1的标准品作为模板,用本试验创新的qRT-PCR方法进行组内、组间重复性测试,利用所得Ct值计算变异系数。如表1所示,组内变异系数为0.45%~1.20%,小于1.50%,组间变异系数为1.14%~1.66%,小于2.0%,表明该方法重复性和稳定性较好。

表1 qRT-PCR重复性试验结果
2.5 牛场环境样品中lsa(E)基因拷贝数量和相对丰度检测结果
运用本试验创新的qRT-PCR方法对采集的24份牛场环境样品进行检测。如表2所示,牛场环境样品中lsa(E)基因拷贝数量的范围为2.54×104~1.52×108copies·μL-1,相对丰度范围为9.39×10-4~1.20×103,说明本方法适用性广。使用普通PCR方法检测牛场环境样品中的lsa(E)基因的存在情况,仅能定性地在NNF1、RCF2、RCF3、NBF2、RDT1、RDT2、RDT3和RDY1 8份环境样品中检出,且8份样本中lsa(E)基因拷贝数量均大于1×106copies·μL-1。

表2 环境样品中lsa(E)基因拷贝数量、相对丰度和lsa(E)基因检出情况
3 结论和讨论
lsa(E)基因不仅位于细菌染色体基因组上,而且也能被质粒携带,同时lsa(E)基因通常与其他耐药基因组合在一起形成不同类型的耐药基因簇。此外,lsa(E)基因在不同来源(人、动物和环境)的细菌中均有报道,且在多个国家广泛分布。同时,lsa(E)基因的广泛存在影响林可胺类、链阳菌素A类和截短侧耳素类等抗生素的治疗效果,对公共卫生安全造成威胁。目前,关于lsa(E)基因在畜禽养殖环境中的存在情况尚不清晰。针对环境中耐药基因的研究方法主要有细菌分离培养技术、qRT-PCR检测技术以及宏基因组学方法,但是养殖环境中绝大多数细菌为不可培养菌,极少量可分离培养的细菌不利于对养殖环境中耐药基因流行分布情况进行全面评价,同时宏基因组学的经济和技术成本都较高。因此,本研究通过创新lsa(E)基因qRT-PCR检测方法,使后续细菌基因组及养殖环境中该基因的检测和分析更加有效。
qRT-PCR检测方法不同于普通PCR,通过向反应体系中加入SYBR荧光基团,只有嵌入在双链DNA小沟的空隙中的荧光基团才能被激发出荧光,因此反应过程中目的基因和荧光强度的增加是对应的。对反应过程中的荧光强度进行监测,不仅能够明确目的基因在反应过程中的数量变化,而且还可以推断模板中目的基因的数量。与普通PCR相比,qRT-PCR具有便捷、快速、准确的优势,该技术目前应用广泛,尤其是在病原微生物的诊断、基因拷贝数量和表达量检测等方面[14-15,20]。
本研究通过创新lsa(E)基因qRT-PCR的检测方法,不仅能够检测纯化培养的细菌基因组中的lsa(E)基因,而且也可检测土壤、粪便等环境样本中lsa(E)基因的拷贝数量和相对丰度。本研究表明,该检测方法熔解曲线均为平滑单峰,特异性强;扩增效率良好101.56%;标准曲线呈现良好的线性关系(r2≥0.997);检测拷贝数量范围广(1.71×102~1.71×108copies·μL-1)且灵敏度高。同时,该检测方法的标准曲线r2值与其他研究中耐药基因(blaKPC、blaNDM和blaOXA-48)qRT-PCR检测方法的标准曲线的r2值(0.995~0.997)相当[21],但高于耐药基因(optrA、cfr和poxtA)qRT-PCR检测方法的标准曲线的r2值(0.992、0.994和0.995)[19],且相邻稀释梯度的扩增曲线在增长的对数期间隔也基本一致。该检测方法的检测范围与BONTRON等[22]报道的mcr-1的标准曲线所适用的检测范围相比,范围更广,说明本试验设计的qRT-PCR引物和程序条件均较好。此外,使用该检测方法对牛场采集的24份环境样本中lsa(E)基因的拷贝数量和相对丰度进行测定,结果显示lsa(E)基因拷贝数量的范围为2.54×104~1.52×108copies·μL-1,相对丰度的范围为9.39×10-4~1.20×103,其中土壤样品RDT2中lsa(E)基因的相对丰度最高,为1.20×103。使用普通PCR方法检测这些样品中lsa(E)基因,仅能在lsa(E)基因拷贝数量大于1×106copies·μL-1的样品中定性地检测到,而采用qRT-PCR检测方法灵敏度达到了100 copies·μL-1,且普通PCR检测方法还需要与测序技术相结合,费时费力。本研究创新了多重耐药基因lsa(E)的qRT-PCR检测方法,特异性强,灵敏度高,检测范围广,且能定量检测于不同养殖环境样品(粪便、土壤、淤泥等)中lsa(E)基因,为lsa(E)基因流行和传播的监测提供技术支持。
