1例发作性运动障碍患儿的GNAO1基因变异研究及文献分析
2023-09-01周珍胡文静吴丽文廖红梅王苗肖政辉
周珍,胡文静,吴丽文,廖红梅,王苗,肖政辉
运动障碍疾病是一组神经系统疾病,又称锥体外系疾病,是一组以不自主动作、动作缺失或缓慢而无瘫痪、姿势及肌张力异常等运动症状为主要表现的神经系统疾病;通常由基底神经节或其连接功能的改变引起。运动障碍的病因主要有脑内核团变性、脑内神经网络异常、遗传基因异常等。随着分子诊断技术的发展,越来越多遗传性病因被认识。近年来研究发现,GNAO1基因突变除导致早发性婴儿癫痫性脑病外,也可导致运动障碍。其关联的运动障碍疾病诊断为神经发育障碍伴非自主运动(neurodevelopmental disorder with involuntary movements,NEDIM),主要临床特征有全面性发育迟缓、肌张力障碍、舞蹈症、运动障碍,以及小头畸形、癫痫发作、胼胝体发育不全、巨脑室、头部控制差等。该病较为罕见,国内鲜有报道。患者临床表现各异,轻重程度不一。我科收治1 例以发作性运动障碍、发育迟缓为主要临床表现的GNAO1基因变异患儿,报道如下。同时总结其临床表现、血生化、肌电图、头颅MRI 等特点,并复习相关文献,探讨GNAO1基因变异的临床特点及基因型与表型的关系,以提高临床医生对GNAO1基因及运动障碍疾病的认识。
1 病例与方法
1.1 临床资料
1.1.1 病例资料 患儿,男,3岁2个月,主因“间发不自主活动2 年余”于2020 年10 月8 日入院。2 年余前,患儿呼吸道感染后出现口面运动障碍,睡眠后消失,持续约5~7 d,感染控制后病情好转,未行特殊处理。1 年余前,患儿患急性阑尾炎后出现口面运动障碍,肢体不自主活动,持续约10余天,予抗感染、镇静处理后好转。5 d前,患急性化脓性阑尾炎,后出现口面运动障碍,肢体不自主活动;外院就诊全麻下行阑尾切除术,上述症状加重,予镇静处理后不能缓解,遂转入我院。个人史:第1 胎第1产,41 周无发作剖宫产,出生体质量3.8 kg,5 个月能抬头,1 岁半能独坐,目前不能爬、扶走、说话;家族史、既往史无特殊。体格检查:生命体征平稳,营养状况欠佳。头围49.5 cm,体质量13.5 kg;神清、精神反应差,能完成父母简单指令。皮肤巩膜(-),肝脾未扪及,四肢肌力、肌张力减低,膝反射未引出,病理征阴性。辅助检查:血铅、铜蓝蛋白、血氨、血乳酸、血氨基酸+酰基肉碱谱、尿有机酸检测:未见明显异常。脑脊液常规+生化:正常;脑电图示:异常小儿脑电图,背景节律明显慢化,异常活动同步脑电图无异常放电;双下肢肌电图:未见异常;头颅MRI:双侧额颞部脑外间隙增宽,幕上脑室稍大,脑沟裂增宽加深;全脊髓MRI:未见异常。诊疗经过:入院后予抗感染、咪达唑仑持续镇静,仍有明显不自主活动;予左旋多巴口服2 d,患儿不自主活动明显好转。家属自觉患儿食欲减差,症状明显缓解,自行停用,感染控制后出院。出院半月随访,无异常动作;随访至2021 年12 月,间有呼吸道感染,暂无发作性运动障碍。1.1.2 二代测序 患儿家属签署知情同意书后,分别抽取患儿及父母外周血(EDTA 抗凝管)2 mL,送至北京全谱医学检验实验室测序,二代测序结果经一代测序验证。
基因测序结果显示患儿16号染色体的GNAO1基因第6外显子存在c.626G>A新发突变,母亲、父亲均为野生型,见图1。该位点是已被报道的致病突变[1];蛋白软件预测结果,Provean危害性预测为deleterious (-4.72),mutationtaster 危害性预测为disease_causing (1),Polyphen2_HVAR 危害性预测为probably damaging(1.0),SIFT 危害性预测为damaging(0.0),提示该突变为致病突变。GNAO1基因与NEDIM(OMIM:617493)相关,为常染色体显性遗传,该患者的GNAO1基因携带致病的杂合突变,翻译时将精氨酸置换为组氨酸,影响异三聚体鸟嘌呤结合蛋白的活性,导致疾病的发生。
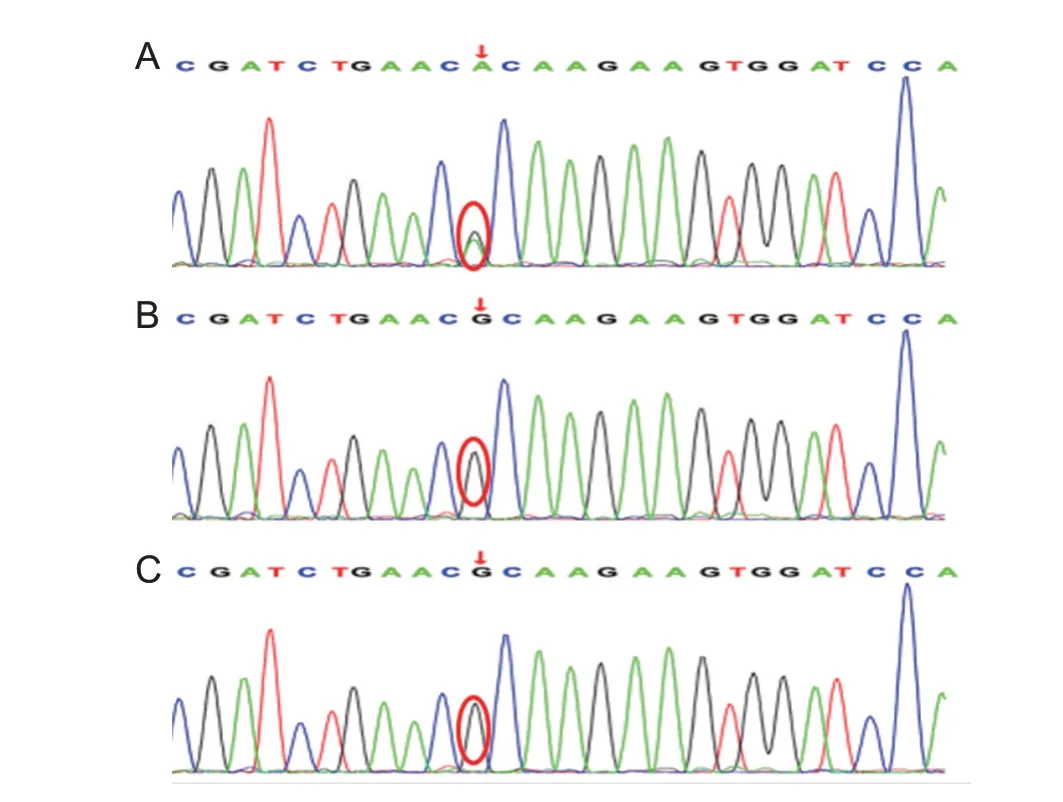
图1 患儿及父母基因测序结果
1.2 方法
过在万方数据库、中国知网、PubMed 数据库,以“GNAO1、神经发育障碍伴非自主运动”、“GNAO1、neurodevelopmental disorder with involuntary movements”为检索词,收集所有相关文献,检索时间截止2022年12月。结合本病例患儿,对其临床资料进行分析。
2 结果
检索符合条件的中文文献0 篇,英文文献6 篇[2-7]。共15 例NEDIM患者携带GNAO1基因突变,结合本例患儿共有16例。对其基因突变类型及临床资料进行分析。发现16例NEDIM患者,男9例,女7例。共携带9个GNAO1突变,其中6个错义突变,2个框内缺失和1 个剪接位点突变。错义突变包括:p.Arg209His(3 例)、p.Glu237Lys(2 例)、p.Arg209Cys(1 例)、p.Glu246Lys(2 例)、p.Ala227Val(1 例)、p.Arg206Leu(1 例);框内缺失包括:p.Ala338del(1 例)、p.Ala301del(1 例);剪接位点突变包括:c.724-8G>A(4例)。87.5%(4/16)有运动障碍、81.3%(13/16)有肌张力障碍、62.5%(10/16)有舞蹈症、37.5%(6/16)有刻板动作、100%(16/16)有发育迟滞。
3 讨论
运动障碍通常由基底神经节或其连接的功能改变引起,随着分子诊断技术的发展,越来越多遗传性病因被认识。2013年Nakamura 等[8]首次在癫痫脑病患儿发现GNAO1新发杂合突变,定义由该类基因突变所致癫痫脑病为早发型癫痫脑病17型(developmental and epileptic encephalopathy 17,EIEE17)。近年来研究发现,GNAO1基因突变具有表型异质性,不同位点突变临床表型不同,除早发癫痫脑病,也可表现为不伴有癫痫发作的NEDIM临床表型。
GNAO1基因位于16q13,编码异三聚体鸟嘌呤结合蛋白(G蛋白)α亚基多肽链O(Gαo),G蛋白是一个庞大的信号转导分子家族,由α、β和γ亚基组成。G蛋白家族成员最广泛的特征是鸟嘌呤核苷酸结合蛋白α亚基,能够水解GTP,并与特定的受体和效应分子相互作用。Gαo在脑组织中表达十分丰富,可以构成高达0.5%的膜蛋白[9],尤其是在海马、纹状体及小脑表达最多[10]。Gαo可耦合α2肾上腺素能、多巴胺D2和5-羟色胺等多种重要的受体,而这些受体在神经递质的释放及运动、神经发育中起关键性的作用[11]。1998年Jiang等[12]通过动物实验发现,敲除GNAO1基因的小鼠临床表型复杂,可出现全身性震颤、癫痫、严重的运动控制障碍、异常行为、痛觉过敏等神经系统症状并早期死亡。Kehrl等[13]发现GNAO1基因G184S杂合突变的小鼠在围产期或生命早期死亡,其与严重的癫痫发作和(或)发作间期癫痫样放电所致的猝死相关,而纯合突变小鼠基本不能存活。另外G α o mRNA 可调节离子通道,在电生理试验中发现GNAO1突变导致其介导的钙电流抑制下降[8]。
目前报道的GNAO1基因突变有20余种,大多为错义突变,另也有框内缺失和剪接位点突变的相关报道[11]。GNAO1基因不同位点变异临床表型不同,其主要关联早发型癫痫脑病17型、NEDIM2种临床表型,遗传方式为常染色显性遗传。研究发现G蛋白功能丧失或功能减低与癫痫发作相关,而功能获得或正常功能突变更多的在有运动障碍伴或不伴癫痫发作的患儿中发现[14]。目前报道的与运动障碍相关的GNAO1基因变异位点主要包括R209 H/L/G/C、E246K、G203R、E237K 等[11]。NEDIM主要临床表现有肌张力障碍、舞蹈症、口面运动障碍、非自主运动、发育迟缓、运动障碍、运动过度等,其次是肌阵挛、痉挛、言语缺失、脑萎缩/大脑萎缩、胼胝体发育不全、巨脑室、头部控制差、小头畸形等。Kulkarni 等[11]报道了两兄弟均患有NEDIM,通过全外显子组测序发现其均为新发错义突变,考虑其父母之一生殖细胞嵌合体可能。Saitsu 等[15]报道2 名无关联的NEDIM 患者,GNAO1基因均为新发突变。Anadth 等[16]报道的6 例NEDIM 患者,其中包括2 名同胞,有4 例患者突变位点是E246K,1例患者为R209H,另1例患者为R209G,所有患者均无癫痫发作,提示这些突变可能是运动障碍所特有的。文献报道46 例GNAO1相关运动障碍患者的临床特征,主要表现为舞蹈症(58.7%)、肌张力障碍(65.2%)、运动障碍(63%)、运动障碍急剧恶化(45.7%)[17]。据文献报道,早期以运动障碍为主要表现的患者,可到10岁后才出现癫痫发作[18,19]。
本病例以感染、手术等打击诱发反复发作性肢体不自主活动、口面运动障碍、肌张力障碍,伴有发育迟滞为主要表现,无癫痫发作。基因检测结果提示为GNAO1基因R209H的杂合新发突变,符合常染色体显性遗传疾病发病机制,结合患儿临床表现及既往文献报道,符合GNAO1基因突变所致的NEDIM临床表型,先证者及其家系成员表型及基因型符合家系共分离。
文献报道在运动障碍患者中,丁苯那嗪是最常用且最有效的药物,因其对囊泡单胺转运蛋白2(vesicular monoamine transporter 2,VMAT2)的作用可耗竭多种胺类神经递质(多巴胺、去甲肾上腺素和5-羟色胺),导致Go信号通路传导广泛减少(如:多巴胺受体、肾上腺素能受体和5-羟色胺受体)[16,20]。相关文献表明左乙拉西坦[11]、托吡酯[15]、苯海索[21]均有效,而托吡酯在抑制携带p.R209C 突变患者的舞蹈症方面非常有效[22];部分病例报告提示苯巴比妥、苯二氮卓类药物、左旋多巴、部分抗精神病药(氟哌啶醇、利培酮)和抗癫痫药(卡马西平、丙戊酸)对过度运动的控制均有不同程度的效果[16,22-25]。对于药物治疗效果不佳患者,进行苍白球深部脑电刺激(DBS)反应都很好,并且非自主运动均得到了控制[19,26];另外也有报道行苍白球切开术可以部分改善[18];但没有报道评估DBS在其他大脑区域(如丘脑下核)的有效性。
本例患儿予口服左旋多巴2 d 后症状明显好转,结合以往文献报道,提示该患儿口服左旋多巴有效,如再次出现发作性运动障碍可及早使用,同时注意避免感染、手术等打击,必要时可行深部脑刺激(DBS)手术治疗。国内外仅有发作性运动障碍病例报道极少,也印证不同的突变位点可致不同的临床表型,而且即使是同一突变位点同种临床表型,其临床轻重程度不一。这也提示我们当一个致病基因可能引起不同种疾病表型时,要充分考虑表型异质性等特殊情况。对于颅内无明显器质性病变,但存在运动障碍、发育迟滞的患儿,建议尽早行基因检测进一步明确病因。
