融合关键反应段特征信息的代谢路径排序方法
2023-08-29谢雨丝卢吉晓黄毅然
谢雨丝,卢吉晓,黄毅然,钟 诚
1(广西大学 计算机与电子信息学院,南宁 530004)
2(广西医科大学附属肿瘤医院 放疗技术中心,南宁 530021)
1 引 言
代谢路径功能上是细胞内参与特定功能的一组反应或酶,给定起始化合物和目标化合物,代谢路径预测是使用计算方法在代谢数据库中预测和分析从起始化合物到目标化合物的新的替代路径[1,2].人们可以通过这些替代路径发现具有应用价值的新的合成代谢路径[3,4].
代谢路径预测方法寻找新替代路径的一种主要方式是以化合物为节点,以连接化合物的代谢反应为边构建代谢网络图,并在代谢网络图中搜索连接起始化合物与目标化合物的路径[5,6].由于化合物和代谢反应种类繁多,由它们构成的代谢网络规模巨大,代谢路径预测方法在代谢网络中两节点间可能会找到数以千计的结果路径[7,8],因此需要通过合适的代谢路径排序方法对返回的结果路径进行排序,以使用户可以从数量众多的路径搜索结果中快速找到感兴趣且具有生化相关性的替代路径[9].
现有的代谢路径预测方法通过选取代谢路径的相关局部特征作为权重来为路径排序.一些代谢路径预测方法[10]通过代谢反应的吉布斯自由能判断路径中的化学反应是否热力学可行,并结合路径中的反应自由能对结果路径进行排序以获取热力学可行性好的替代路径[11].例如,代谢路径预测方法Phdseeker利用结果路径中的代谢反应的吉布斯自由能为每一个代谢反应评分,将所有代谢反应评分汇总值作为结果路径排序的因素[12].除了路径中代谢反应的吉布斯自由能特性,化合物对宿主的毒性可能会影响代谢路径的产物产量,因此也常作为路径排序的一个因素.例如,Carbonell等人设计的代谢路径预测方法XTMS将结果路径中的每个中间底物对宿主的毒性汇总,并用毒性汇总值作为结果路径排序的依据[13].代谢反应数量少的代谢路径在代谢工程实践中通常比代谢反应数量多的路径更容易合成.为此,Ravikrishnan等人设计的代谢路径预测方法Metquest采用代谢路径的长度作为路径排序因素,以选取代谢路径中的反应数量相对较少的替代路径[7].
此外,通过追踪由起始化合物转移到目标化合物的原子或原子团来寻找代谢路径能够有效避免簇化合物[14].一些代谢路径预测方法利用代谢路径中从起始化合物到目标产物间的原子或原子团的转移数量对代谢路径进行排序,以获得生化相关性较好的路径.例如,代谢路径预测方法BPAT-M[15]和Retrace[16]利用代谢路径长度、代谢路径中化合物间的碳原子转移数量来为代谢路径排序.BPFinder[17]和AGPathFinder[18]方法则结合反应底物与产物的分子结构相似程度、路径中化合物间的原子团转移数量来为代谢路径排序.
代谢路径的生化相关性主要体现在路径中的反应段表现出的特定功能,如糖酵解路径通过将葡萄糖转化为丙酮酸释放和吸收能量[19],这种功能性路径由特定的反应段构成,在代谢网络中经常出现[20].虽然现有代谢路径预测方法通过利用代谢路径的局部生化特征,可以准确选取满足特定需求的代谢路径,但是通过路径的局部特征对结果路径排序没有考虑多个化合物之间及多个代谢反应段之间的相互作用和联系,不易于发现具有结构功能特征的代谢路径.针对这些问题,本文提出了一种新的代谢路径排序方法KPRank以提高代谢路径预测结果质量,主要贡献如下:
1)提出代谢路径关键反应段的概念,以捕捉和度量代谢路径的结构功能特征.
2)提出一种融合代谢路径关键反应段的信息熵与互信息对代谢路径进行评分和排序的模型,以充分利用代谢路径的结构功能特征信息.
在KEGG代谢数据库[21-23]上的实验结果表明,通过KPRank方法对代谢路径预测算法Retrace,BPFinder和MetQuest的路径预测结果进行排序可以发现生化相关性更好的代谢路径,有效提高了代谢路径预测结果的质量.
2 方 法
2.1 问题定义
代谢路径排序,即对代谢路径预测算法的路径搜索结果中的代谢路径按路径生化相关性特征或用户感兴趣的特征对路径进行量化评分和排序,为用户快速准确提供生化相关性好的替代路径.代谢路径排序方法之间的差异主要是为路径排序所选取的路径特征不同,以及路径特征的量化评分方式的不同.在代谢路径排序中选择合适的路径排序特征,设计合理的路径特征的量化评分方法,是准确地发现生化相关的替代路径的关键.
2.2 已知代谢路径集合构建
本文通过KEGG的PATHWAY数据库[24]的67个已知代谢网络来建立已知代谢路径集合P,以便从已知代谢路径集合P中选取合适的路径排序特征对代谢路径进行排序.
对于一个已知代谢网络,首先枚举该代谢网络的所有化合物的成对组合,以每个化合物对组合中的两个化合物分别作为起始化合物与目标化合物,在这个代谢网络里采用Yen等人设计的最短路径搜索算法[25]搜索连接这对化合物的前5条线性路径,然后将所有找到的线性路径保存到已知代谢路径集合P里.接下来通过同样的方式从67个已知代谢网络中的每一个代谢网络中提取线性路径并保存到已知代谢路径集合P里,并去除已知代谢路径集合P中重复的路径.
图1展示了已知代谢路径集合构建过程的一个例子,其中cn1、cn2、cn3和cn4是代谢网络rn1中的化合物节点,R20~R25是连接化合物节点的代谢反应.
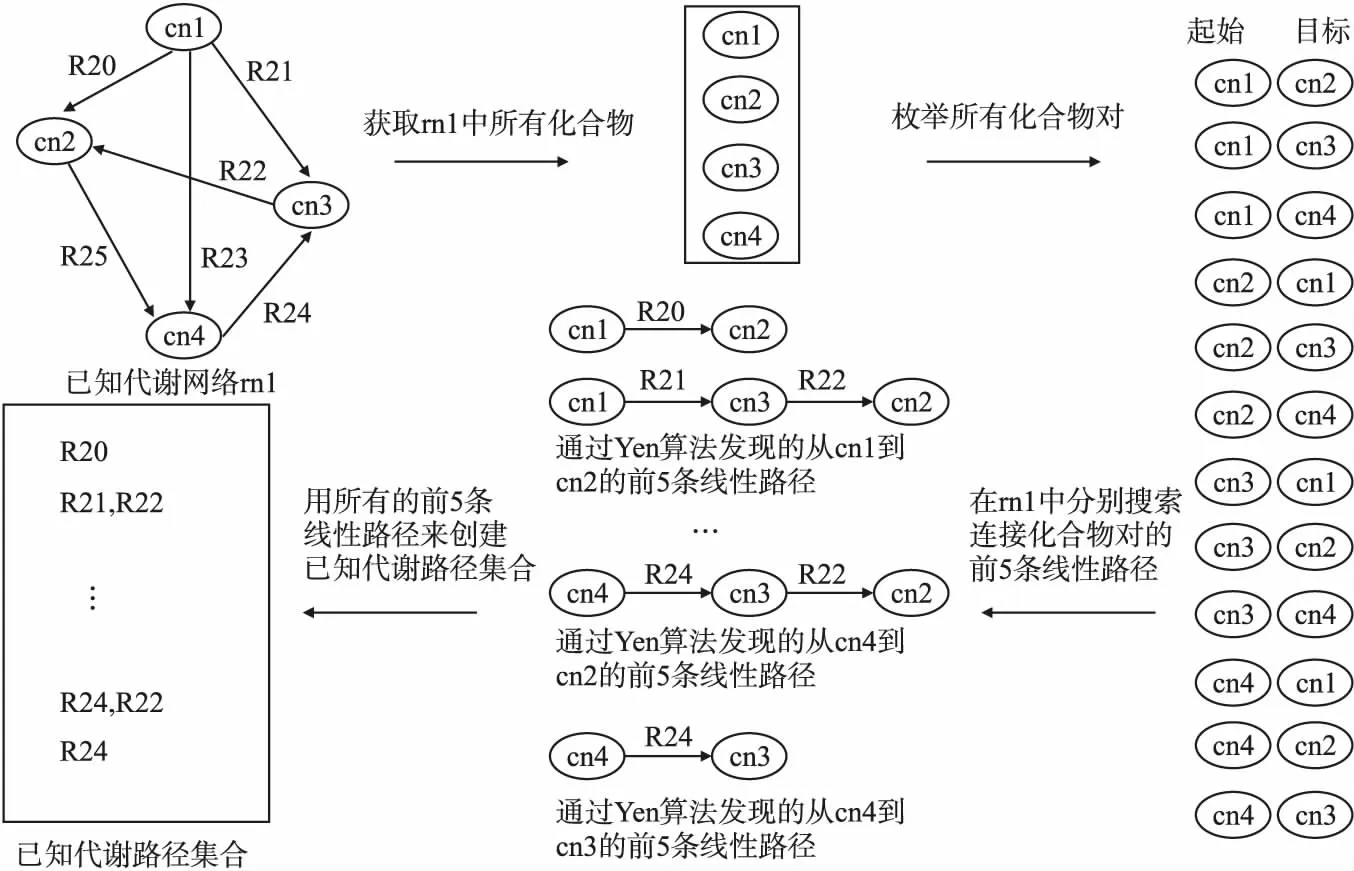
图1 构建已知代谢路径集合示例
由图1可以看到,在构建已知代谢路径集合时,首先选出代谢网络rn1的所有化合物cn1、cn2、cn3和cn4,枚举它们的成对组合,并以每对化合物组合中的两个化合物分别作为起始化合物和目标化合物,然后在代谢网络rn1中通过Yen算法[25]搜索连接这对化合物的前5条线性路径,最后将找到的线性路径存入已知代谢路径集合,同时去掉重复的代谢路径.
2.3 关键反应段
特定功能路径是表现出特定生化功能的连续固定的代谢反应组合,它们通常大量出现在不同物种的代谢网络中[20].例如,三羧酸循环路径是各种需氧生物体内普遍存在的连接糖类、脂类及氨基酸类化合物的代谢路径[19].为更充分的挖掘路径反应的结构功能特征,本文提出代谢路径关键反应段KRS(Key Reaction Set)概念,以捕捉和度量代谢路径的反应结构功能.
对于已知代谢路径集合P中的每一条代谢路径,每个反应段可由路径中的连续2个、3个或者4个反应构成.可以统计每个反应段在已知代谢路径集合P中出现的次数.关键反应段是在已知代谢路径集合P中出现次数大于100次的反应段.关键反应段通常是已知代谢网络中高频出现的连续反应组合,这些反应组合前后的代谢反应搭配丰富,反应组合间的关联程度较高.
本文引入两个评估关键反应段的指标以评估关键反应段中反应间的关联程度:关键反应段的信息熵和互信息值.信息熵是某条消息所含的信息量,它反映的是获知某个消息后,关于该事件的不确定性的减少量[26].对已知代谢路径集合P,关键反应段的左反应是关键反应段最左边的反应在关键反应段出现的路径p∈P里的前一个反应,关键反应段的右反应是关键反应段最右边的反应在关键反应段出现的路径p∈P里的后一个反应.设Se是由m个连续反应组成的关键反应段,L是关键反应段Se的左反应集合,Y是关键反应段Se的右反应集合,关键反应段Se的左反应信息熵Hl(Se)和右反应信息熵Hy(Se)分别可以通过式(1)和式(2)计算[26]:
Hl(Se)=-∑x∈Lf(x)log2f(x)
(1)
Hy(Se)=-∑y∈Yf(y)log2f(y)
(2)
式(1)和式(2)中的f(x)和f(y)分别是左反应x∈L和右反应y∈Y在已知代谢路径集合P里出现的频率.关键反应段的左、右信息熵越大,说明该关键反应段前后反应搭配越丰富,通过关键反应段信息熵可以评估关键反应段的外部反应搭配的丰富程度.互信息是一种度量离散型随机变量相关程度的方法.为了进一步评估关键反应段内部多个反应之间的相关程度,本文采用改进的互信息计算方法[26]来计算关键反应段的互信息值以评估关键反应段内部反应间的关联程度:
(3)
(4)
(5)
其中MMI(Se)是关键反应段Se的互信息值,Count(Se)是关键反应段Se在已知代谢路径集合P中出现的次数,fr(Se)是关键反应段Se出现在已知代谢路径集合P中的概率,N表示已知代谢路径集合P的路径数量,Avg(Se)是关键反应段Se中不同的反应组合在已知代谢路径集合P中的平均出现概率.通过关键反应段的互信息值MMI(Se)可以反映出关键反应段Se内部多个反应之间的相关程度.关键反应段Se出现在代谢路径集合P中的概率越高,同时关键反应段Se中不同的反应组合在已知代谢路径集合P中出现越少,MMI(Se)也越高,说明关键反应段Se内部的反应组合的关联程度也较高.
在文本信息处理中,互信息可以用于句子分词,将句子中的字符视为随机变量,利用字符在文本句子中出现的概率计算该字符与前后字符之间的互信息值来度量这些字符之间的关联程度[27].类似地,在本文的代谢路径的反应关联分析中,将代谢路径中的代谢反应视为随机变量,通过利用反应在代谢路径集合中的代谢路径里出现的概率计算该反应与前后反应之间的互信息值来度量这些反应之间的关联程度.文本信息处理和代谢反应关联分析中的互信息计算,都是在特定数据集内根据随机变量的出现概率来度量随机变量之间的关联程度,与随机变量的具体形式是字符或代谢反应实际上并没有关系.
2.4 基于关键反应段的代谢路径评分方法
综合关键反应段Se的信息熵和互信息值,可以得到代谢路径的关键反应段Se的评分方法:
Scorekr(Se)=H(Se)+I(Se)
(6)
(7)
(8)
其中Score_kr(Se)是关键反应段Se的评分.H(Se)是关键反应段Se的左信息熵和右信息熵的综合值,其中Hl(Se)是通过式(1)计算得到的关键反应段Se的左信息熵,Hy(Se)是通过式(2)计算得到的关键反应段Se的右信息熵.minHl表示已知代谢路径集合P的所有关键反应段的左信息熵的最小值,maxHl表示已知代谢路径集合P的所有关键反应段的左信息熵的最大值.minHy表示已知代谢路径集合P的所有关键反应段的右信息熵的最小值,maxHy表示已知代谢路径集合P的所有关键反应段的右信息熵的最大值.I(Se)表示关键反应段Se的归一化互信息值,其中minMMI是已知代谢路径集合P的所有关键反应段的互信息值的最小值,maxMMI是已知代谢路径集合P的所有关键反应段的互信息值的最大值.
式(6)结合代谢路径关键反应段的信息熵和互信息来计算关键反应段评分以评估关键反应段的反应搭配的丰富程度和反应间的关联程度,式(6)的评分越高说明关键反应段的反应搭配越丰富,反应间的关联程度越高,生化相关性越好.代谢路径的关键反应段的信息熵与互信息值之间存在一定差别.为均衡评估这两个路径特征对关键反应段评分的作用,本文分别采用式(7)和式(8)对代谢路径的关键反应段的信息熵和互信息值做归一化处理.
基于线性路径pa的所有关键反应段ks,线性路径pa的评分可以通过式(9)计算:
(9)
其中ad为关键反应段权重,其取值范围为[0,1];npa为线性路径pa的长度,ap为线性路径pa的长度权重.在式(9)中,线性路径pa的所有关键反应段评分之和越高、路径长度越短,说明线性路径pa的生化相关性越好.
基于代谢路径PB的每条线性路径pi的评分Score′(pi),代谢路径PB的评分可由式(10)计算得出:
(10)
其中lb是构成代谢路径PB的线性分支路径个数,pi表示构成代谢路径PB的一条线性路径.最后KPRank方法通过式(10)计算代谢路径PB的所有线性分支路径评分的平均值得到代谢路径PB的最终评分.
2.5 算法描述
对于待排序代谢路径集PR,使用本文方法KPRank对待排序代谢路径集合PR中的路径进行排序的基本步骤如下:
Step 1.对已知代谢路径集合P中每一条线性路径,枚举其中由2至4个连续反应构成的反应段,计算每个反应段rs在已知代谢路径集合P中的出现次数,将出现次数大于100次的关键反应段存入关键反应段集合HRS.
Step 2.对于待排序路径集PR中的每一条代谢路径pl中的每条线性分支路径pa,使用逆向最长匹配算法[28]从路径pa中提取反应段,并保留路径pa中属于HRS集合的关键反应段,通过式(6)计算路径pa中的属于HRS集合的每个关键反应段评分,并通过式(9)得到线性路径pa的评分.
Step 3.利用Step 2得到的代谢路径pl包含的所有线性路径的评分,采用式(10)计算出代谢路径pl的评分.
Step 4.重复Step 2和Step 3计算待排序代谢路径集合PR中的每条代谢路径的评分.最后根据PR中的每条路径的评分以从高到低的顺序输出路径排序结果.
算法1形式描述了本文提出的方法KPRank.
算法1.KPRank
输入:已知代谢路径集合P,待排序路径集合PR
输出:待排序路径集合PR的路径排序结果
Begin
for allpathinPdo
RS←枚举路径path中由2至4个连续反应构成的反应段;
end for
for allrsinRSdo
统计反应段rs在P中出现次数;
HRS←在已知代谢路径集合P中出现次数>100的反应段;
end for
for allplinPRdo /*待排序路径集合PR中的每条代谢路径pl*/
for allpainpldo /*代谢路径pl中的每条线性分支路径pa*/
采用逆向最长匹配算法从路径pa中提取属于HRS集合的关键反应段;
利用式(6)计算路径pa的每个关键反应段评分;
基于路径pa的每个关键反应段评分,通过式(9)计算得到线性路径pa的评分;
end for
基于代谢路径pl包含的每条线性路径评分,采用式(10)计算出代谢路径pl的评分;
end for
以待排序路径集合PR中的路径评分降序输出路径排序结果;
End.
3 实 验
3.1 实验环境
为测试KPRank的路径排序效果,本文使用KEGG PATHWAY数据库中的67个已知代谢网络来构建已知代谢路径集合P.
BPFinder是基于原子团追踪的代谢路径预测算法[17],ReTrace是基于原子追踪的代谢路径预测算法[16],MetQuest是基于动态规划的代谢路径预测算法[7].本文分别使用3种预测代谢路径的方法Retrace,BPFinder和MetQuest在KEGG PATHWAY数据库中搜索26条已知代谢路径.分别从https://www.cs.helsinki.fi/group/sysfys/software/retrace/下载了Retrace,从https://github.com/RamanLab/metquest下载了MetQuest.在实验中,每个代谢路径预测方法寻找每条已知路径的起始化合物和目标化合物之间的代谢路径,并返回路径搜索结果的前5条路径,然后利用KPRank方法分别为Retrace,BPFinder和MetQuest找到的代谢路径排序,并分别对经过KPRank排序后和排序前的代谢路径与已知代谢路径进行比较来评估路径排序结果的质量.采用Java语言编程实现KPRank算法.KPRank的实验参数配置为:ad与ap都是0.5.方法Retrace、BPFinder和MetQuest均采用默认运行参数.实验的计算机配置为Intel(R)Xeon(R)CPU 6130 @ 2.10GHz和40GB RAM.
3.2 评估指标
通过计算工具得到的替代路径的生化相关性会影响替代路径在实际生物合成应用的可用性.人们一般通过比较已知路径与计算路径来评估计算路径的生化相关性.本文通过比较计算路径与已知路径得到的7个指标来评估计算路径的生化相关性[29]:
1)最大连通子图边数占比(RELCCS)是已知路径与计算路径同构的子图里最大的联通子图的边数与已知路径的边数之比[29].RELCCS值越大,表明计算路径与已知路径越相似,也说明计算路径具有更好的生化相关性.RELCCS的平均值的计算如下[29]:
(11)
N为计算路径的个数,RELCCSi指第i条计算路径与对应这条计算路径的已知路径同构的子图里最大的连通子图边数与已知路径边数之比[29].
2)化合物灵敏度Sn=tp/(tp+fn),真阳性tp是在计算路径与已知路径中同时出现的化合物,且这些化合物在已知路径中的出现顺序与它们在计算路径中的出现顺序一样[12,18].假阴性fn是在计算路径中不存在,但在已知的路径中出现的化合物[12,18].
3)化合物阳性预测值PPV=tp/(tp+fp),假阳性fp是在计算路径中存在但在已知路径中不存在的化合物[12,18].
4)化合物准确率AC=(SN+PPV)/2,AC值越大,说明算法具有更好的恢复已知路径的化合物的能力[12,18].
5)反应灵敏度R_Sn=r_tp/(r_tp+r_fn),真阳性r_tp是在已知路径和计算路径中同时出现的反应,并且这些反应在已知路径中的出现顺序与在计算路径中的出现顺序一样.假阴性r_fn是在计算路径中不存在,但在已知路径中存在的反应[12,18].
6)反应阳性预测值R_PPV=r_tp/(r_tp+r_fp),假阳性r_fp是在计算路径中存在,但在已知路径中不存在的反应[12,18].
7)反应准确率R_AC=(R_SN+R_PPV)/2,R_AC值越大,说明算法具有更好的恢复已知路径反应的能力[12,18].
3.3 计算路径和已知路径结构的比较
表1显示了各种方法寻找26条已知路径所找到的代谢路径的平均RELCCS值.其中KPRank(BPFinder)、KPRank(Retrace)和KPRank(MetQuest)分别表示用KPRank方法对BPFinder、Retrace、MetQuest方法所找到的代谢路径进行排序以后得到的结果.
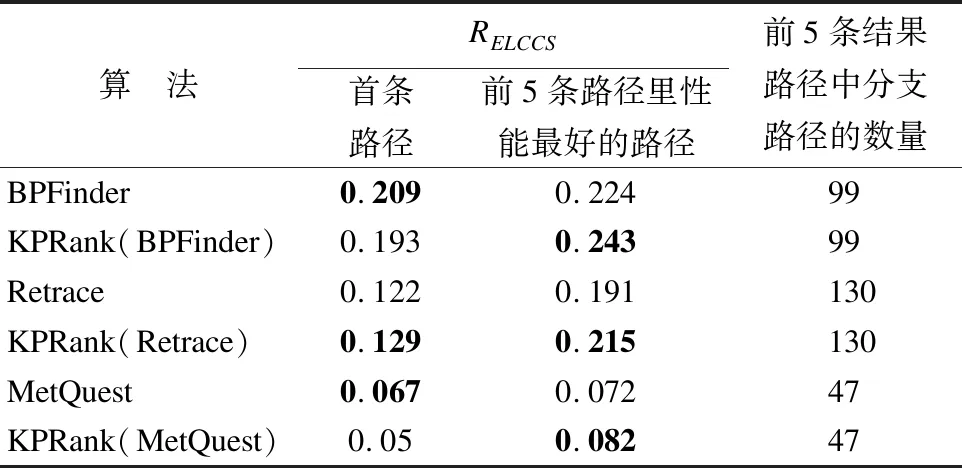
表1 26条计算路径的平均RELCCS值和前5条结果路径中分支路径的数量
由表1可知,对于方法BPFinder、Retrace、MetQuest所找到的前5条路径里性能最好的路径,使用方法KPRank为结果路径排序后得到的路径的RELCCS值皆优于未使用方法KPRank排序得到的路径的RELCCS值.这些结果表明经过方法KPRank对路径结果进一步排序后,所得到的路径结构更接近已知路径,路径搜索质量得到进一步提高.另一方面,从表1可以看到,经过执行方法KPRank排序后,Retrace的首条路径和前5条路径里性能最好的路径的RELCCS值均比原来的方法更好,而BPFinder和MetQuest的首条路径的RELCCS值有一定程度的减少.结合找到的分支路径数量来看,相比其他方法,Retrace找到的前5条路径的分支路径数量更多.这说明方法KPRank更适用于协助分析和寻找分支多、反应组合复杂的分支替代路径.与此同时,从表1还可以看到,3种方法的首条路径的RELCCS值都不是前5条路径里最好的,这表明要寻找拓扑结构上更接近已知路径的替代路径,首条路径不一定是最好的选择.
3.4 计算路径和已知路径中的化合物和反应的比较
本文采用AC,SN和PPV来度量计算路径的化合物精度、敏感度和阳性预测值,采用R_AC,R_SN和R_PPV来度量计算路径的反应精度、敏感度和阳性预测值.表2给出了各种方法的26条计算路径的反应的平均精度AC、平均敏感度SN和平均阳性预测值PPV.

表2 26条计算路径的反应平均R_SN值、平均R_PPV值和平均R_AC值
由表2可以看到,对于BPFinder和Retrace的前5条路径里性能最好的路径,使用方法KPRank为结果路径排序后得到的路径的R_Sn值,R_PPV值和R_AC值都比未使用方法KPRank排序得到的路径的R_Sn值,R_PPV值和R_AC值更好.对于BPFinder的首条路径,经过方法KPRank为结果路径排序后得到的路径的R_PPV值和R_AC值也比没有使用方法KPRank排序得到的路径的R_PPV值和R_AC值更好.对于MetQuest的首条路径和前5条路径里性能最好的路径,没有采用方法KPRank排序得到的路径的R_PPV值和R_AC值相对更好一些.这些结果说明KPRank更适用于BPFinder和Retrace,KPRank对BPFinder和Retrace的路径结果进一步排序可以搜索出路径的反应组成更接近已知路径的替代路径.
表3给出了各种方法的26条计算路径的化合物的平均精度AC、平均敏感度SN和平均阳性预测值PPV.
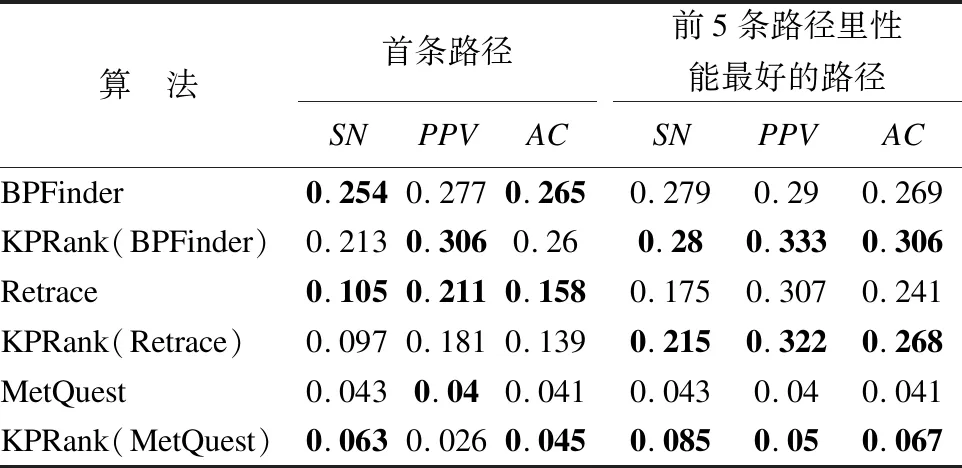
表3 26条计算路径的化合物平均SN值、平均PPV值和平均AC值
从表3可以看到,经过方法KPRank排序后,虽然BPFinder和Retrace的首条路径的Sn值和AC值相对有所下降,MetQuest的首条路径的Sn值和AC值比未使用方法KPRank排序得到的路径的Sn值和AC值好一些.相对于首条路径,前5条路径里性能最好的路径更有代表性,对于方法BPFinder、Retrace、MetQuest所找到的前5条路径里性能最好的路径,采用方法KPRank为结果路径排序后得到的路径的Sn值、PPV值和AC值都好于未使用方法KPRank排序得到的路径的Sn值,PPV值和AC值.这些结果表明经过方法KPRank对这些路径预测方法的路径搜索结果进一步排序后,所得到的路径中的化合物更接近已知路径中的化合物,搜索得到的路径具有更好的还原已知代谢路径的化合物的能力.
表1~表3的结果说明KPRank排序方法能够提升代谢路径预测方法搜索路径的生化相关性,可以有效提高代谢路径预测结果的质量.
3.5 实例分析:磷酸戊糖合成路径的搜索
上述实验结果表明基于路径关键反应段的路径排序方法KPRank有效的提高了代谢路径预测方法的路径搜索结果的生化相关性.本文通过讨论分析一个利用方法KPRank对BPFinder的路径搜索结果经过排序以后得到的路径实例来进一步了解方法KPRank的特点.
5-磷酸核糖(D-Ribose 5-phosphate)是核苷酸和核酸合成所必需的底物,它可以通过磷酸戊糖合成路径中非氧化阶段由β-D-果糖6-磷酸(beta-D-Fructose 6-phosphate)为起始化合物通过代谢合成[30].磷酸戊糖路径存在于细胞的胞浆中,它同时是葡萄糖氧化的替代途径[31],并可用于生产NADPH和5-磷酸核糖[30].
图2(a)是KEGG数据库的已知磷酸戊糖合成路径,图2(b)是使用方法KPRank对BPFinder的磷酸戊糖合成路径的路径搜索结果排序后得到的首条路径,图2(c)是BPFinder搜索磷酸戊糖合成路径得到的首条路径.在图2中,虚线椭圆中的化合物表示代谢路径中的分支化合物,计算路径中与已知路径相同的反应和化合物均以斜体加粗表示.
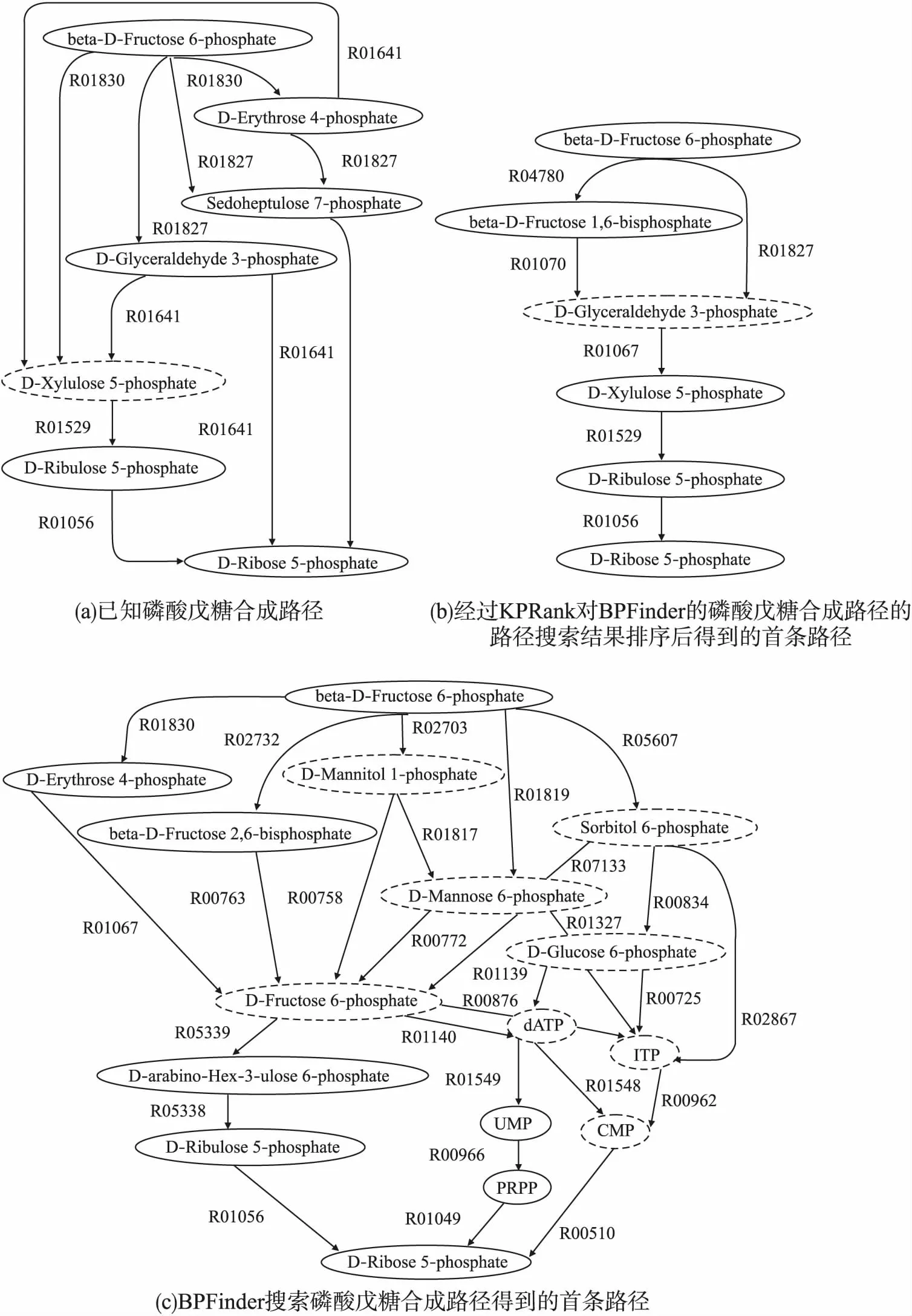
图2 磷酸戊糖合成路径
从图2(a)可以看到反应R01529和R01056构成的反应段是已知磷酸戊糖合成路径的一部分.基于式(6)的计算,由反应R01529和R01056构成的反应段的关键反应段评分是0.99,这说明这个反应段在从67个已知代谢网络中提取的已知代谢路径集合里出现频率较高,反应结构功能特征明显,因此在图2(b)中,KPRank将包含这个反应段的路径作为首条路径输出.有趣的是,图2(b)的这条经过KPRank排序后得到的首条磷酸戊糖合成路径位于未经KPRank排序的BPFinder发现的磷酸戊糖合成路径的第13位,这表明基于关键反应段的评分策略可以更快速有效的发现包含功能反应段的路径.
对比图2(a)和图2(c)可以看到,虽然BPFinder搜索磷酸戊糖合成路径得到的首条路径中有部分化合物和反应出现在已知路径里,但是BPFinder的这条从起始化合物beta-D-Fructose 6-phosphate到目标化合物D-Ribose 5-phosphate的路径分支众多、跨度大,有很大部分与已知路径不同,这在一定程度上可能会增加设计实现新的磷酸戊糖合成路径的难度.对比图2(a)和图2(b)还可以看到,KPRank发现了已知路径中没有的从起始化合物beta-D-Fructose 6-phosphate开始,经过D-Glyceraldehyde 3-phosphate最终到达目标化合物D-Ribose 5-phosphate的新替代路径,同时这条新替代路径中从D-Glyceraldehyde 3-phosphate开始到目标化合物D-Ribose 5-phosphate之间的大部分路径与已知路径相同,这将在一定程度上降低设计实现新的磷酸戊糖合成路径的难度.
4 结束语
本文提出了的代谢路径排序方法的特色是:首先从67个已知的代谢网络提取高频关键反应段,并融合代谢路径的关键反应段的信息熵与互信息对代谢路径预测算法找到的路径进行评分和排序,以获取生化相关性好的替代路径.实验结果表明,利用KPRank对代谢路径预测算法的结果路径进行排序可以找出生化相关性更好的路径,进一步提高了预测的结果路径的质量.
下一步工作将考虑利用代谢数据库的反应规则对已有代谢网络中的代谢反应进行功能性分类,并结合关键反应段和反应功能分类对路径预测结果进行排序,以为代谢路径的分析和合成提供更丰富的替代路径选择.
