线粒体翻译延伸因子Ts对心肌肥大的作用及机制
2023-08-26张德玉王菲高岩岩
张德玉 王菲 高岩岩


[摘要]目的探究線粒体翻译延伸因子Ts(EF-Ts)通过影响线粒体损伤调控病理性心肌细胞肥大的分子机制。方法使用血管紧张素Ⅱ(AngⅡ)制备小鼠心肌肥大的细胞模型与动物模型,采用蛋白免疫印迹(WB)方法检测心肌肥大时EF-Ts的蛋白表达。用EF-Ts干扰慢病毒干扰小鼠原代心肌细胞中EF-Ts的表达,同时用AngⅡ处理,借助四甲基罗丹明甲酯(TMRM)与钙黄绿素乙酰氧基甲酯(Calcein AM)染色,应用激光共聚焦显微镜观察心肌细胞线粒体膜电位和线粒体通透性转换孔(MPTP)水平,并采用WB法检测心肌肥大标志物心房钠尿因子(ANF)和钠尿肽B(NPPB)的变化情况。结果心肌肥大的细胞模型和动物模型中EF-Ts蛋白表达量均明显降低(t=2.95、14.93,P<0.05)。与对照组相比,AngⅡ诱导的心肌肥大细胞模型中TMRM和Calcein AM染色荧光强度均明显减弱,在心肌肥大细胞模型中敲低EF-Ts后TMRM和Calcein AM染色荧光强度进一步减弱(F=4.22~22.88,P<0.05),而心肌肥大标志物ANF、NPPB蛋白表达量进一步上升(F=6.52、20.96,P<0.05)。结论EF-Ts缺失可能引起线粒体功能损伤而导致心肌肥大,因此EF-Ts有可能成为干预心肌肥大的重要靶标。
[关键词]肽链延伸,翻译;线粒体;肌细胞,心脏;肥大;小鼠
[中图分类号]R394;R541[文献标志码]A[文章编号]2096-5532(2023)03-0401-06
doi:10.11712/jms.2096-5532.2023.59.108[开放科学(资源服务)标识码(OSID)]
[网络出版]https://link.cnki.net/urlid/37.1517.R.20230809.1714.001;2023-08-1013:49:21
ROLE OF MITOCHONDRIAL TRANSLATION ELONGATION FACTOR TS IN CARDIAC HYPERTROPHY AND UNDERLYING MECHANISM ZHANG Deyu, WANG Fei, GAO Yanyan (Institute of Translational Medicine, Qingdao University, Qingdao 266071, China)
[ABSTRACT]ObjectiveTo explore the molecular mechanism of the mitochondrial translation elongation factor EF-Ts re-gulating pathological cardiomyocyte hypertrophy through affecting mitochondrial injury. MethodsAngiotensin Ⅱ (AngⅡ) was used to prepare cell and animal models of cardiac hypertrophy in mice. Western blot was used to measure the expression of EF-Ts protein in cardiac hypertrophy. EF-Ts-interfering lentivirus was used to interfere the expression of EF-Ts in mouse primary cardiomyocytes. After treatment with Ang Ⅱ and staining with tetramethylrhodamine methyl ester (TMRM) and calcein acetoxymethyl ester (Calcein AM), a laser confocal microscope was used to observe the mitochondrial membrane potential and mitochondrial permeability transition pore level of cardiomyocytes. Western blot was used to determine the changes of atrial natriuretic factor (ANF) and natriuretic peptide B (NPPB), which were the markers of cardiac hypertrophy. ResultsThe expression of EF-Ts protein in both the cell model and animal model of cardiac hypertrophy was decreased significantly (t=2.95,14.93;P<0.05). Compared with those in the control group, the fluorescence intensities of TMRM and Calcein AM were significantly decreased in Ang Ⅱ-induced cell model of cardiac hypertrophy, and were further significantly reduced after knocking down EF-Ts in the model of cardiac hypertrophy (F=4.22-22.88,P<0.05), while the expression of ANF and NPPB proteins was further significantly increased (F=6.52,20.96;P<0.05). ConclusionEF-Ts deletion may cause mitochondrial dysfunction, and thereby lead to cardiac hypertrophy. Therefore, EF-Ts may be a key target for the intervention of cardiac hypertrophy.
[KEY WORDS]peptide chain elongation, translational; mitochondria; myocytes, cardiac; hypertrophy; mice
心肌肥厚是指心脏为适应各类刺激而出现的心肌细胞体积和质量增大的病症[1]。心肌肥厚形成是一个慢性且复杂的过程,是许多心血管疾病的病理生理基础[1-5],但其分子机制依然不明确。线粒体在心肌细胞中发挥重要功能,包括ATP生成、活性氧(ROS)产生、代谢调控、钙平衡等。研究发现,线粒体功能障碍(MD)与心肌肥厚、高血压等诸多心血管疾病存在联系[6]。线粒体代谢动力学失衡、钙稳态失衡、ROS水平升高、线粒体DNA受损均对心肌肥厚的形成、发展起到一定影响[7],因此MD也是心肌肥厚形成、发展的一个关键影响因素[8-9]。而线粒体翻译的正常有序进行,是线粒体功能的重要保
402青岛大学学报(医学版)59卷
证。线粒体翻译是一个动态平衡的过程,这一过程受到多种因子的调控。线粒体翻译可以分为起始、延伸、终止3个阶段[10],其中延伸阶段在蛋白质合成方面发挥着关键作用,被视作保守性最强的翻译阶段[11]。当前已知的线粒体翻译延伸因子(EF)包括EF-Tu、EF-Ts和EF-G1,通过其表达量的变化能够掌握线粒体的功能与翻译速度,同时能够对细胞功能状态做出有效评估,这些因子突变定然会引发线粒体病变[11-13]。EF-Ts是一种核编码的线粒体蛋白。有研究采用全外显子测序方法在严重的心肌病病人体内鉴定了TSFM(EF-Ts的编码基因)的突变[14-16]。这提示EF-Ts的正常表达对维持正常的心肌细胞功能有重要作用,然而关于EF-Ts在心肌肥厚中的作用迄今并无太多研究报道。因此,本研究从病理性心肌肥大的细胞与动物模型入手,探讨心肌细胞肥大对EF-Ts表达的影响,以及干预EF-Ts对线粒体功能的作用,为心肌肥厚的临床研究提供新的靶点与思路。
1材料与方法
1.1实验材料
日龄1~2 d的C57小鼠乳鼠、C57小鼠(大任富城),血管紧张素Ⅱ(AngⅡ,Abmole),DMEM/F12(SparkJade),胎牛血清(BI),青链霉素(美仑生物),胰液素、Ⅱ型胶原酶和TRITC Phalloidin罗丹明标记鬼笔环肽(翊圣生物),四甲基罗丹明甲酯(TMRM) Perchlorate(Abmole),线粒体通透性转换孔(MPTP)检测试剂盒(碧云天),EF-Ts干扰慢病毒和HitransG P(吉凯基因),冰台、眼科剪、弯头镊、小烧杯、260目过滤网和生理盐水(美仑生物),心房钠尿因子(ANF)抗体和EF-Ts抗体(Abcam),钠尿肽B(NPPB)抗体(Affinity),Gapdh抗体(CST)。
1.2实验方法
1.2.1小鼠原代心肌细胞的分离和培养于冰台上准备3个6 cm培养皿,各加入4 mL PBS。取雌雄不分的1~2 d日龄C57小鼠乳鼠放入一容器中,用体积分数0.75的乙醇清洗小鼠1~2次。于超净台中用眼科剪将乳鼠心脏取出,放入盛有PBS的培养皿中,洗3次,用吸管吸掉PBS。将心脏剪碎后加入消化液,转移到灭菌的50 mL离心管中。37 ℃摇动水浴,每次5 min,将上层液体转移至小烧杯中,再次加入消化液摇动水浴,反复10次以上直到消化完毕。转移烧杯內液体到50 mL离心管内,接着再添加4 mL血清,使消化结束。收集消化产物,以800 r/min离心5 min,弃上清,收集沉淀,再用5 mL DMEM/F12轻轻将沉淀悬浮,再以800 r/min离心5 min,弃上清,收集沉淀。在沉淀中加入10 mL含有体积分数0.05血清的DMEM/F12培养液充分重悬,用260目过滤网(先用1 mL培养液润湿)过滤,将过滤后的液体收集到10 cm培养皿中,置于细胞培养箱内于37 ℃下进行细胞培养。细胞差速贴壁1.0~1.5 h后,收集细胞悬液,此时没有贴壁的细胞即为心肌细胞。最后将细胞接种于适合的细胞培养皿中,进行后续实验。
1.2.2心肌肥大细胞模型的构建与鉴定分离的小鼠原代心肌细胞培养24 h后,用DMEM/F12无血清培养液预处理24 h,将培养液更换为含体积分数0.10血清的DMEM/F12培养液。实验分2组,其中实验组加入1.5 μmol/L的AngⅡ,对照组加入同等体积的PBS,处理48 h后提取细胞总蛋白,进行WB检测。
1.2.3心肌肥大动物模型的构建与鉴定将小鼠称质量后分为2组,每天于同一时间点,实验组腹腔注射1.5 mg/kg的Ang Ⅱ(生理盐水配制),对照组注射等体积的生理盐水,2周后两组腹腔注射30 g/L的水合氯醛,待小鼠彻底麻醉后断颈处死取出心脏。取部分心脏组织加入RIPA裂解液(每20 mg组织加入150 μL)于组织研磨器中充分研磨后,在冰上裂解1 h(每10 min涡旋震荡10 s),4 ℃下以12 000 r/min离心15 min,收集上清用于WB检测。
1.2.4肥大相关蛋白的WB检测将处理好的细胞用冰冷的PBS漂洗2次,加入RIPA裂解液(含1×PMSF和1×cooktail)在冰上裂解30 min(每10 min涡旋震荡10 s),4 ℃下以12 000 r/min离心15 min。收集上清,加入4×蛋白上样缓冲液,98 ℃煮沸10 min。根据所需检测的蛋白大小选择适合浓度的SDS-PAGE凝胶分离蛋白并转移到PVDF膜上,用50 g/L的脱脂牛奶封闭非特异性结合位点1 h后,将膜和按比例稀释的一抗4 ℃孵育过夜,以TBST洗膜3次(每次10 min)后,用抗鼠或者抗兔二抗孵育1.5 h,再次以TBST洗膜3次(每次10 min),最后用ECL显影液显影并通过化学发光仪检测信号,以Gapdh为参照分析条带灰度。实验重复3次。
1.2.5细胞病毒感染当心肌细胞生长到密度为70%~80%时,即可以进行EF-Ts慢病毒感染,以multiplicity of infection(MOI)=15的病毒量感染细胞,同时根据培养皿大小加入适量促感染试剂HitransG P,24 h后更换培养液。细胞感染72 h收集细胞用于后续实验。
1.2.6心肌细胞线粒体膜电位检测将原代心肌细胞接种于35 mm玻底培养皿中,并将细胞分为空白对照组(A组)、Ang Ⅱ处理组(B组)、NC+Ang Ⅱ处理组(C组)和shEF-Ts+Ang Ⅱ处理组(D组)。病毒感染处理同1.2.5,Ang Ⅱ处理同1.2.2。将培养液吸除干净,用PBS沿壁清洗细胞1或2次。在1 mL无血清培养液中加入150 nmol/L的TMRM,与细胞37 ℃共孵育60 min。用PBS溶液清洗1次,借助激光共聚焦显微镜观察荧光强度的变化,TMRM的激发波长和发射波长分别为543、580 nm。实验重复3次。
1.2.7心肌细胞MPTP测定将原代心肌细胞接种于35 mm的玻底培养皿中,细胞分组及处理同1.2.6。MPTP测定严格按照MPTP试剂盒说明书进行操作。Calcein AM的激发波长和发射波长分别为494、517 nm。实验重复3次。
1.3统计学方法
应用软件GraphPad Prism 5进行统计分析。计量数据结果以±s表示,两组比较采用t检验,多组比较采用单因素方差分析,以P<0.05为差异有统计学意义。
2结果
2.1心肌肥大模型的建立及EF-Ts蛋白表达
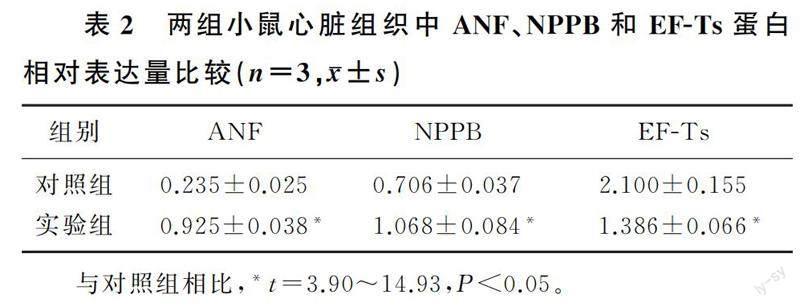
与对照组相比,心肌肥大细胞模型的指标蛋白ANF、NPPB的表达量明显升高,EF-Ts蛋白的表达量明显降低(t=2.95~4.01,P<0.05)。在心肌肥大动物模型中,同样能检测到ANF、NPPB的表达量明显升高,EF-Ts的表达量明显降低(t=3.90~14.93,P<0.05)。见图1和表1、2。
2.2各组心肌细胞线粒体膜电位比较
与空白对照组比较,Ang Ⅱ处理组荧光强度大幅减弱,表明线粒体膜电位下降,线粒体膜出现损伤,伤及线粒体膜功能;敲低EF-Ts后线粒体膜电位进一步降低,表明线粒体膜功能受损更加严重。见图2。空白对照组、Ang Ⅱ处理组、NC+Ang Ⅱ处理组和shEF-Ts+Ang Ⅱ处理组的平均荧光强度分别为124.200±4.575、92.580±3.868、94.130±5.152和74.520±3.400(n=3),差异具有统计学意义(F=22.88,P<0.05)。
2.3各组心肌细胞MPTP的比较
与空白对照组相比,Ang Ⅱ处理组的荧光强度在5和10 min时均有所减弱,敲低EF-Ts后,荧光强度降低更加明显(F=4.22、14.03,P<0.05)。表明敲低EF-Ts加重了Ang Ⅱ所造成的MPTP开放和线粒体膜通透性增强。见图3、表3。
2.4EF-Ts敲低后肥大心肌细胞中肥大标记物ANF和NPPB的变化
与空白对照组比较,Ang Ⅱ处理组心肌细胞中心肌肥大标记物ANF和NPPB的蛋白表达量明显升高,使用慢病毒敲低EF-Ts后其表达量进一步上升(F=6.52、20.96,P<0.05),表明敲低EF-Ts有一定的促心肌肥大的作用。见图4、表4。
3讨论
肥厚型心肌病被发现已有60多年,该病是具有高猝死风险的遗传性疾病,其临床表型多样[17-20]。心肌肥厚的发生发展涉及多种化学物质和信号通路[20]。心脏作为机体代谢最活跃的器官,线粒体的密度非常高,它参与心肌细胞代谢过程中的能量生成、信号传导、ROS生成、细胞凋亡等活动,在维持正常的心脏功能中发挥着重要作用[21-26]。有研究发现,在心肌肥厚的发生发展进程中,线粒体呼吸链复合体的活性降低、ATP合成能力下降[27-28]、钙稳态失衡以及ROS水平升高都是线粒体功能紊乱的重要表现[8]。既往有文献报道,MD与心肌肥大、高血压以及心肌缺血灌注损伤有关[29-30]。线粒体能量代谢、氧化应激以及线粒体参与的钙稳态等都与心肌肥大的发生和发展有着密切关联,越来越多的证据表明MD可能是心肌肥厚过程中的一个关键的因素[31]。所以,将MD作为心肌肥厚治疗及预防的靶点具有极高的价值。
正常心肌产生的ATP中95%以上来自线粒体的氧化磷酸化。有研究表明,在心肌肥大过程中,线粒体最大氧化能力下降的部分原因是呼吸链复合物的活性降低,线粒体膜电位下降和ATP合成酶活性降低影响ATP的产生[32]。心肌肥大向心力衰竭转变过程中便伴随着线粒体功能的衰退及氧化磷酸化能力的降低。EF-Ts参与线粒体基因组编码的13个多肽的翻译[33-34],这13个多肽都是线粒体呼吸链复合体中的核心组分[35-36]。EF-Ts的正常表达对维持线粒体电子传递链功能和氧化磷酸化水平的正常至关重要[11,37]。有研究显示,TMSF突变造成的EF-Ts缺陷会导致肥厚型、扩张型心肌病和脑心肌病的发生,部分肥厚型心肌病病人线粒体复合体Ⅰ、Ⅳ和Ⅴ活性显著降低,线粒体翻译障碍、功能受损,这表明EF-Ts缺陷有可能是通过影响线粒体功能导致心肌肥大[14,16,38]。近年有研究报告了1例TSFM基因中新的复合杂合子变体,病人的组织样本中EF-Ts蛋白大量减少,线粒体复合体Ⅰ和Ⅳ活性降低,线粒体翻译功能受损,造成严重的线粒体功能紊乱,导致线粒体心肌病[15],从而确认TSFM是心脏功能障碍的主要靶点。
本研究结果显示,Ang Ⅱ诱导的心肌肥大的细胞与动物模型中,EF-Ts蛋白表达量明显降低,而在心肌肥大细胞模型中干扰EF-Ts表达,线粒体膜电位受损和MPTP開放程度以及肥大标志物ANF、NPPB的表达量进一步增加。这表明EF-Ts通过影响线粒体功能对心肌细胞的肥大过程造成影响,EF-Ts缺失可能引起线粒体功能损伤而导致心肌肥大。在后续工作中我们将进一步探究EF-Ts在心肌细胞肥大过程中的具体调控通路,从而为心血管疾病的预防和治疗提供新的思路和靶标。
[參考文献]
[1]NAKAMURA M, SADOSHIMA J. Mechanisms of physiolo-gical and pathological cardiac hypertrophy[J]. Nature Reviews Cardiology, 2018,15(7):387-407.
[2]GIBB A A, HILL B G. Metabolic coordination of physiological and pathological cardiac remodeling[J]. Circulation Research, 2018,123(1):107-128.
[3]LI Y Q, LIANG Y J, ZHU Y J, et al. Noncoding RNAs in cardiac hypertrophy[J]. Journal of Cardiovascular Translatio-nal Research, 2018,11(6):439-449.
[4]MCMULLEN J R, JENNINGS G L. Differences between pathological and physiological cardiac hypertrophy: novel the-rapeutic strategies to treat heart failure[J]. Clinical and Expe-rimental Pharmacology & Physiology, 2007,34(4):255-262.
[5]THAM Y K, BERNARDO B C, OOI J Y, et al. Pathophy-siology of cardiac hypertrophy and heart failure: signaling pathways and novel therapeutic targets[J]. Archives of Toxicology, 2015,89(9):1401-1438.
[6]惠汝太. 肥厚型心肌病是一个最早能够看见解决答案的心血管疾病[J]? 中国分子心脏病学杂志, 2018,18(2):2393-2395.
[7]WALTERS J W, AMOS D, RAY K, et al. Mitochondrial re-dox status as a target for cardiovascular disease[J]. Current Opinion in Pharmacology, 2016,27:50-55.
[8]胡欢,李萍,程晓曙. 线粒体功能障碍与心肌肥厚的研究进展[J]. 重庆医学, 2018,47(23):3081-3083.
[9]ZHOU L Y, LIU J P, WANG K, et al. Mitochondrial function in cardiac hypertrophy[J]. International Journal of Car-diology, 2013,167(4):1118-1125.
[10]DESAI N, YANG H T, CHANDRASEKARAN V, et al. Elongational stalling activates mitoribosome-associated quality control[J]. Science (New York, N Y), 2020,370(6520):1105-1110.
[11]OTT M, AMUNTS A, BROWN A. Organization and regulation of mitochondrial protein synthesis[J]. Annual Review of Biochemistry, 2016,85:77-101.
[12]SCHULZ C, SCHENDZIELORZ A, REHLING P. Unlocking the presequence import pathway[J]. Trends in Cell Biology, 2015,25(5):265-275.
[13]MAI N, CHRZANOWSKA-LIGHTOWLERS Z M A, LIGHTOWLERS R N. The process of mammalian mitochondrial protein synthesis[J]. Cell and Tissue Research, 2017,367(1):5-20.
[14]SMEITINK J A, ELPELEG O, ANTONICKA H, et al. Distinct clinical phenotypes associated with a mutation in the mitochondrial translation elongation factor EFTs[J]. American Journal of Human Genetics, 2006,79(5):869-877.
[15]PERLI E, PISANO A, GLASGOW R I C, et al. Novel compound mutations in the mitochondrial translation elongation factor (TSFM) gene cause severe cardiomyopathy with myo-cardial fibro-adipose replacement[J]. Scientific Reports, 2019,9(1):5108.
[16]EMPERADOR S, BAYONA-BAFALUY M P, FERNN-DEZ-MARMIESSE A, et al. Molecular-genetic characterization and rescue of a TSFM mutation causing childhood-onset ataxia and nonobstructive cardiomyopathy[J]. European Journal of Human Genetics, 2017,25(1):153-156.
[17]TEARE D. Asymmetrical hypertrophy of the heart in young adults[J]. British Heart Journal, 1958,20(1):1-8.
[18]HERSHBERGER R E, GIVERTZ M M, HO C Y, et al. Correction: Genetic evaluation of cardiomyopathy: a clinical practice resource of the American College of Medical Genetics and Genomics (ACMG)[J]. Genetics in Medicine: Official Journal of the American College of Medical Genetics, 2019,21(10):2406-2409.
[19]MARIAN A J, BRAUNWALD E. Hypertrophic cardiomyopathy: genetics, pathogenesis, clinical manifestations, diagnosis, and therapy[J]. Circulation Research, 2017,121(7):749-770.
[20]MARIAN A J. Molecular genetic basis of hypertrophic cardiomyopathy[J]. Circulation Research, 2021,128(10):1533-1553.
[21]GOFFART S, VON KLEIST-RETZOW J C, WIESNER R J. Regulation of mitochondrial proliferation in the heart: power-plant failure contributes to cardiac failure in hypertrophy[J]. Cardiovascular Research, 2004,64(2):198-207.
[22]JIANG D S, WEI X, ZHANG X F, et al. IRF8 suppresses pathological cardiac remodelling by inhibiting calcineurin signalling[J]. Nature Communications, 2014,5:3303.
[23]PUTINSKI C, ABDUL-GHANI M, STILES R, et al. Intrinsic-mediated caspase activation is essential for cardiomyocyte hypertrophy[J]. Proceedings of the National Academy of Sciences of the United States of America, 2013,110(43):E4079-E4087.
[24]WENDE A R, ONEILL B T, BUGGER H, et al. Enhanced cardiac Akt/protein kinase B signaling contributes to patholo-gical cardiac hypertrophy in part by impairing mitochondrial function via transcriptional repression of mitochondrion-targeted nuclear genes[J]. Molecular and Cellular Biology, 2015,35(5):831-846.
[25]DWORATZEK E, MAHMOODZADEH S, SCHUBERT C, et al. Sex differences in exercise-induced physiological myocardial hypertrophy are modulated by oestrogen receptor beta[J]. Cardiovascular Research, 2014,102(3):418-428.
[26]ANDERSSON D C, FAUCONNIER J, YAMADA T, et al. Mitochondrial production of reactive oxygen species contri-butes to the β-adrenergic stimulation of mouse cardiomycytes[J]. The Journal of Physiology, 2011,589(Pt 7):1791-1801.
[27]SPERL W, JESINA P, ZEMAN J, et al. Deficiency of mitochondrial ATP synthase of nuclear genetic origin[J]. Neuromuscular Disorders, 2006,16(12):821-829.
[28]ODONNELL J M, FIELDS A, XU X Y, et al. Limited functional and metabolic improvements in hypertrophic and healthy rat heart overexpressing the skeletal muscle isoform of SERCA1 by adenoviral gene transfer in vivo[J]. American Journal of Physiology Heart and Circulatory Physiology, 2008,295(6):H2483-H2494.
[29]FU Y L, TAO L, PENG F H, et al. GJA1-20k attenuates Ang Ⅱ-induced pathological cardiac hypertrophy by regulating gap junction formation and mitochondrial function[J]. Acta Pharmacologica Sinica, 2021,42(4):536-549.
[30]MCDERMOTT-ROE C, YE J M, AHMED R, et al. Endonuclease G is a novel determinant of cardiac hypertrophy and mitochondrial function[J]. Nature, 2011,478(7367):114-118.
[31]WST R C I, DE VRIES H J, WINTJES L T, et al. Mitochondrial complex I dysfunction and altered NAD(P)H kine-tics in rat myocardium in cardiac right ventricular hypertrophy and failure[J]. Cardiovascular Research, 2016,111(4):362-372.
[32]MANN D L, ZIPES D P, LIBBY P, et al. Braunwalds heart disease: a textbook of cardiovascular medicine[M]. Amsterdam: Elsevier, 2015.
[33]LEE D E, BROWN J L, ROSA M E, et al. Translational machinery of mitochondrial mRNA is promoted by physical activity in Western diet-induced obese mice[J]. Acta Physiologica, 2016,218(3):167-177.
[34]MERCER T R, NEPH S, DINGER M E, et al. The human mitochondrial transcriptome[J]. Cell, 2011,146(4):645-658.
[35]YOKOKAWA T, MORI R, SUGA T, et al. Muscle denervation reduces mitochondrial biogenesis and mitochondrial translation factor expression in mice[J]. Biochemical and Biophysical Research Communications, 2020,527(1):146-152.
[36]YOKOKAWA T, KIDO K, SUGA T, et al. Exercise-induced mitochondrial biogenesis coincides with the expression of mitochondrial translation factors in murine skeletal muscle[J]. Physiological Reports, 2018,6(20):e13893.
[37]LIN Y Y, LI F J, HUANG L L, et al. eIF3 associates with 80S ribosomes to promote translation elongation, mitochon-drial homeostasis, and muscle health[J]. Molecular Cell, 2020,79(4):575-587.e7.
[38]SCALA M, BRIGATI G, FIORILLO C, et al. Novel homozygous TSFM pathogenic variant associated with encephalocardiomyopathy with sensorineural hearing loss and peculiar neuroradiologic findings[J]. Neurogenetics, 2019,20(3):165-172.
(本文編辑马伟平)