草铵膦的应用、生物合成与降解
2023-08-25李斌赵昌明
李斌?赵昌明
摘要:传统的非选择性除草剂市场近十几年来发生了剧烈的变化,比如百草枯由于其较高的毒性和致死率逐渐被多个国家禁用,草甘膦由于抗性的累积以及对环境造成的负担市值逐渐降低,草铵膦(phosphinothricin,PT)作为后起之秀市场占比正在稳步增加。草铵膦是20世纪70年代发现的一种次膦酸化合物,可以与谷氨酰胺合成酶结合,不可逆地抑制其活性,从而杀灭杂草。以草铵膦为药效核心的天然产物双丙氨膦生物合成途径中存在许多不寻常的生化反应:首先,大多数非核糖体肽合成酶会选择含有未修饰的α-氨基的底物,而双丙氨膦合成途径中的非核糖体肽合成酶选择了α-氨基被乙酰化修飾的底物;其次,草铵膦结构中独特的C-P-C(碳-磷-碳)键结构单元的生物合成涉及到的P-甲基转移酶既是研究的热点,也是研究的难点。另一方面,作为一种天然产物,草铵膦在生物降解方面具有得天独厚的优势。虽然目前对草铵膦具体的生物降解途径所知甚少,但从其它的膦酸天然产物生物降解的研究中我们可以得到很多有益的启示。本文将从草铵膦的应用、生物合成、生物降解3个方面展开,对草铵膦的研究现状进行综述。
关键词:草铵膦;双丙氨膦;除草剂;非核糖体肽;次膦酸
中图分类号:R978.1文献标志码:A
Application, biosynthesis and degradation of phosphinothricin
Li Bin and Zhao Chang-ming
(School of Pharmaceutical Sciences, Wuhan University, Wuhan 430072)
Abstract The traditional non-selective herbicide market has experienced dramatic changes. For example, paraquat has been gradually banned in many countries for its high toxicity and lethality; the market value of glyphosate is decreasing due to the accumulation of resistance and the burden on the environment; the market share of phosphinothricin is increasing steadily as a rising star. Phosphinothricin is a kind of phosphinic acid compound found in 1970s, which can irreversibly bind glutamine synthetase, inhibit its activity, and accordingly kill weeds. Many interesting reactions happened in the biosynthesis of bialaphos, a nature product with phosphinothricin as the pharmacophore. The majority of nonribosomal peptide synthetases prefer substrates with a free α-amino group, while the nonribosomal peptide synthetase involved in bialaphos biosynthesis takes the N-acetylated one. Characterization of P-methyl transferase responsible for the synthesis of unique C-P-C moiety is not only a research hotspot, but also a grand challenge. Meanwhile, as a natural product, phophinothricin has showed unique advantages in biodegradation. Although the exact biodegradation pathway of phosphinothricin remains unclear, many useful insights can be drawn out from studies on the biodegradation of other phosphonic acid natural products. In this article, the topic will focus on three aspects of phosphinothricin: Application, biosynthesis, and biodegradation.
Key words Phosphinothricin; Bialaphos; Herbicide; Nonribosomal peptide; Phosphinate
1 草铵膦的应用
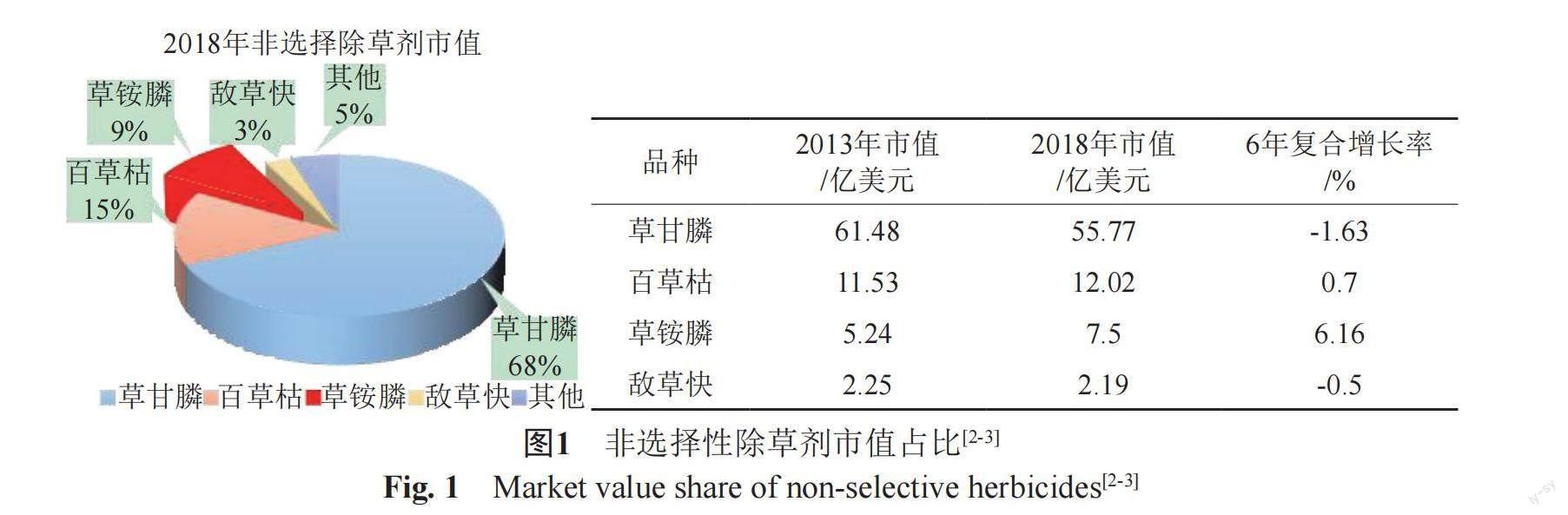
除草剂的使用是现代农业生产中的重要环节,自1941年第一代除草剂2,4-二氯苯氧乙酸(2, 4-D)、2-甲基-4-氯苯氧乙酸(MCPA)问世以来,除草剂迅速给农业化工业和杂草管理带来了变革[1]。自此以后的约40年里,几乎每两到三年都会有新的作用机制的除草剂问世,直到20世纪90年代至今不再有新的作用机制的除草剂出现。目前在非选择性除草剂中,占据市场份额较高主要是草铵膦(1, phosphinothricin, PT或glufosinate ammonium, 商品名保试达(basta)、百速顿等)、草甘膦(2, glyphosate)、百草枯(paraquat)和敌草快(diquat),约占非选择性除草剂市场份额的95%。截至2018年,草甘膦仍然高居除草剂市值榜首,占据非选择性除草剂的68%,百草枯,草铵膦和敌草快次之(图1)[2-3]。2013—2018年,敌草快市值略有下降,占比不高;百草枯的市值虽然略有上升,但是由于较高的毒性和误服致死率,其在多个国家已被禁用,正在逐渐退出市场;剩下的草甘膦和草铵膦表现出较高的竞争力。而作为新兴的除草剂,草铵膦由于其低毒高效、尚未出现较多的耐药性,市场占比正在稳步上升。
草甘膦是一种化学合成的针对5-烯醇式丙酮酰莽草酸合成酶(5-enolpyruvate-3-shikimate phosphate synthase, EPSPS)的高效非选择性除草剂,在通过转基因技术获得抗草甘膦农作物之后,它迅速取代了百草枯、敌草快等成为世界范围内广泛使用的除草剂[4]。然而,近年来草甘膦的使用出现了意料外的状况,它在极低浓度的情况下可以刺激植物的生长(即毒物兴奋作用,hormesis),虽然这并非草甘膦独有的,但是草甘膦表现出更明显也更具有重复性的毒物兴奋作用[5]。另外,草甘膦可以作为抗生素杀灭环境中的微生物[6],从而影响生态环境。随着使用时间的增长,杂草对草甘膦的耐药性不断累积,导致草甘膦的药效降低,为达到除草效果单位面积的施药量不断提高,对环境的危害越来越大,寻找草甘膦的有效替代品成为迫切的需求,此时与草甘膦结构类似的化合物草铵膦进入人们的视线。
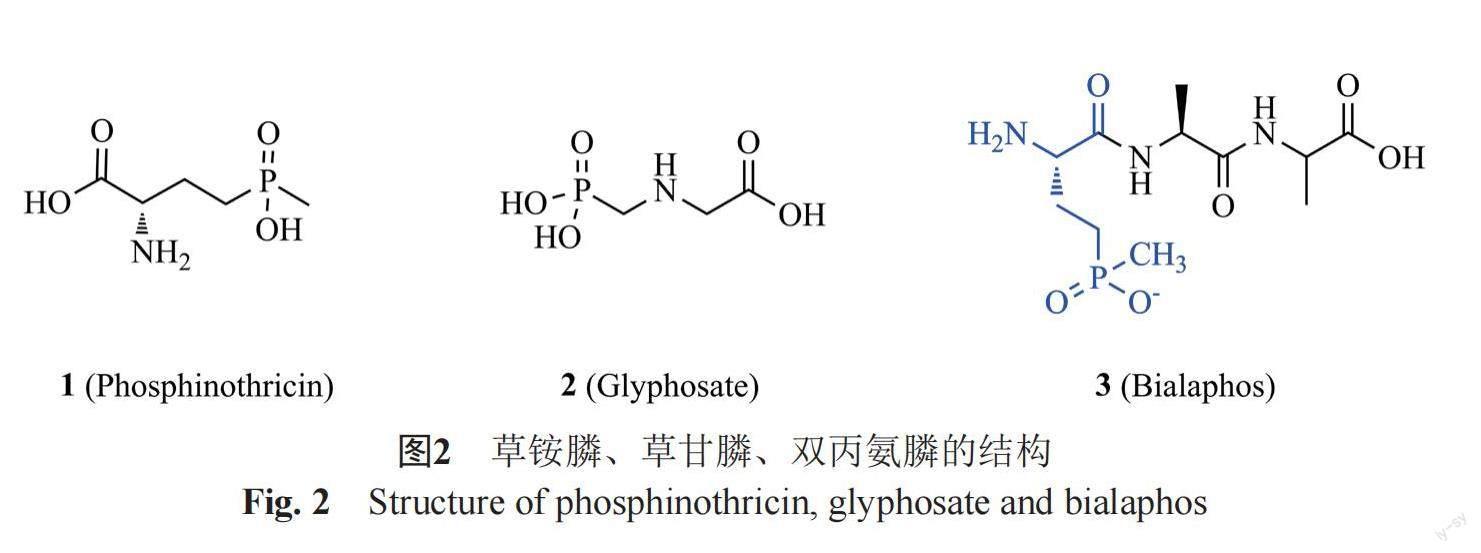
草铵膦是目前发现的唯一一种次膦酸(phosphinic acid)类天然产物,它含有独特的C-P-C结构单元。20世纪70年代发现的由产绿色链霉菌Streptomyces viridochromogenes DSM40736[7]和吸水链霉菌Streptomyces hygroscopicus ATCC21705[8]产生的三肽物质双丙氨膦(3, bialaphos,BA)中含有草铵膦结构单元,不久后在鬼臼孢菌Kitasatosporia phosalacinea DSM 43860中发现的PT-Ala-Leu三肽[9-10]以及在吸水链霉菌KSB-1285中发现的PT-Ala-Ala-Ala四肽[11]中同样含有此结构单元。草铵膦、草甘膦、双丙氨膦的结构见图2。
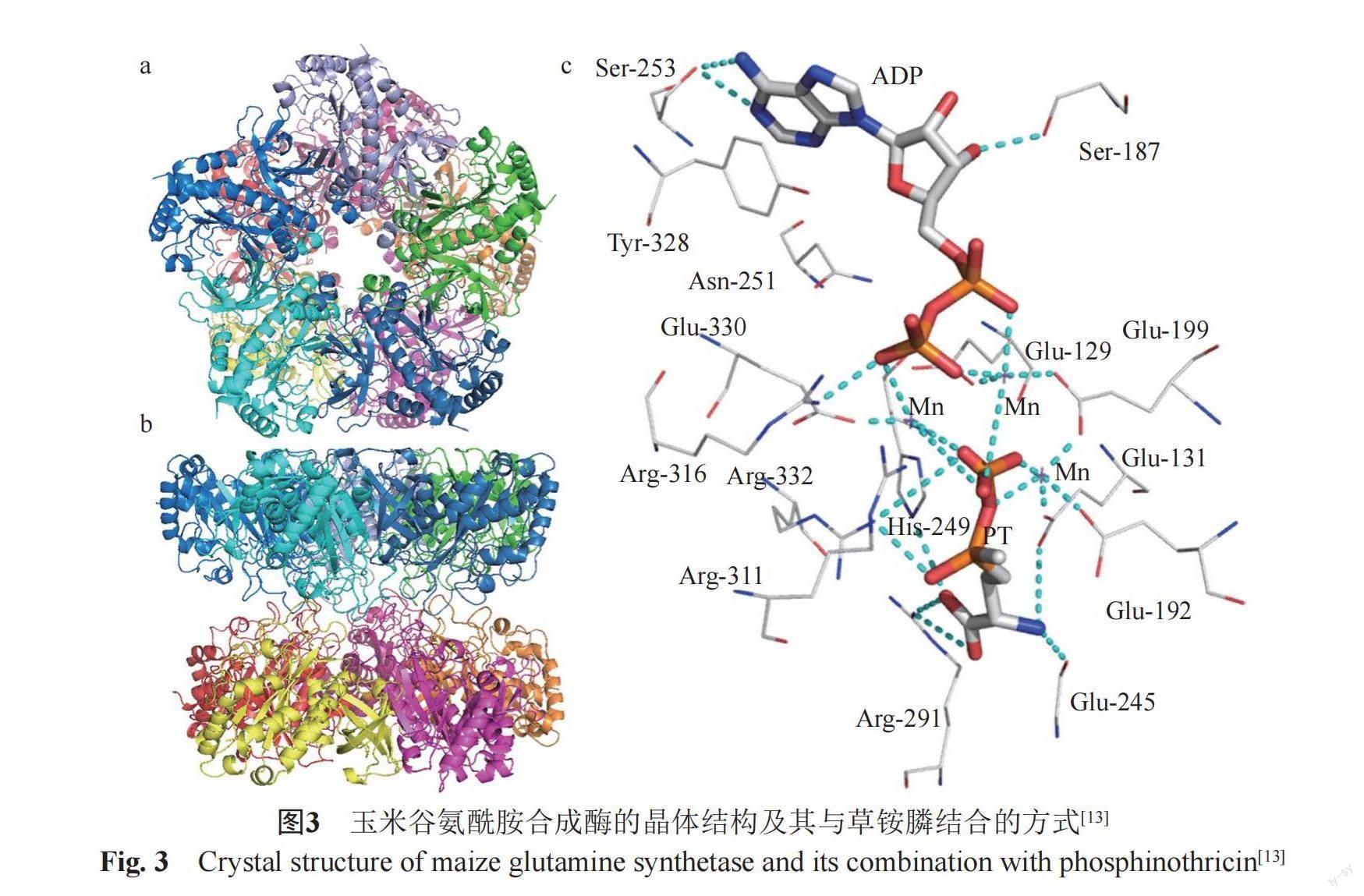
草铵膦是1种ATP依赖的谷氨酰胺合成酶(glutamine synthase, GS)抑制剂。谷氨酰胺合成酶在生物体内发挥着重要的生理功能,它催化游离铵与谷氨酸进行反应产生谷氨酰胺,从而解除由硝酸盐还原、光呼吸作用产生的游离铵带来的毒性。草铵膦在进入植物体内之后会与ATP结合,占据谷氨酰胺合成酶的活性位点,从而不可逆地抑制谷氨酰胺合成酶的活性。谷氨酰胺合成酶的活性被抑制会导致植物体内的氮代谢紊乱,游离铵无法被代谢而过量累积,从而引起叶绿素失活,最终导致植物体的死亡[12-13]。玉米中谷氨酰胺合成酶(PDB: 2D3C)是由两个对称的环状五聚体形成的十聚体结构(图3a~b),组成两个五聚体的亚基是相同的,五聚体内每两个亚基连接处的残基形成一个活性位点,共有10个活性位点。草铵膦与ADP、Mn2+形成复合体,复合体中ADP的结合位点位于亚基连接处裂缝的开口;草铵膦与玉米谷氨酰胺合成酶的结合位点在裂缝的底部,主要通过氢键相互作用与主链上的Gly-245以及侧链上的Glu-131, Glu-192, His-249, Arg-291, Arg-311, 和Arg-332残基结合,膦酸基团则与3个Mn2+结合(图3c)[14]。
草铵膦抑制谷氨酰胺合成酶活性的特性被用来进行除草剂的开发,早期主要用于葡萄、柑橘及其它水果果园中。由于草铵膦的作用特点,需要避免草铵膦产品与作物的绿色部分接触,因此必须直接喷洒在作物枝干以下,这也是限制其适用范围的一种因素。
双丙氨膦生物合成途径的乙酰化修饰酶pat/bar基因负责对中间体的氨基进行修饰,其编码的蛋白也可以修饰草铵膦的α-氨基,使其不能发挥活性。
1987年,Block等[15]成功将pat/bar基因转入烟草、马铃薯、番茄细胞中,獲得了对草铵膦除草剂具有抗性的作物。此后,草铵膦作为一种新型的除草剂进入市场,凭借其低毒高效、易降解无残留等优势逐渐占领市场。然而,目前主流的草铵膦合成方法均为化学方法,合成原材料昂贵、工艺复杂,对设备要求较高且易产生有毒有害废弃物[16],这大大提高了草铵膦的生产成本,很大程度上限制了草铵膦的使用。解析草铵膦及含草铵膦化合物的生物合成途径,寻找绿色、高效的生物合成工艺制备草铵膦,是降低草铵膦生产成本的有效途径。
2 草铵膦的生物合成
本文以含草铵膦结构单元的化合物——双丙氨膦的生物合成为例介绍草铵膦的生物合成途径。产绿色链霉菌DSM 40736(php 基因簇)[17-18]和吸水链霉菌ATCC 21705(bcp 基因簇)[19]中合成双丙氨膦的基因簇的基因序列几乎完全相同。为方便表述,本文中统一使用php基因簇进行指代。双丙氨膦的生物途径合成主要可以分为3个阶段(图4):首先是双丙氨膦骨架亚磷酸(18)的形成。随后,由pat/bar基因编码的乙酰转移酶对氨基进行修饰,并由非核糖体肽合成酶(nonribosomal peptide synthetases,NRPS)引入两个丙氨酸形成三肽(20)。最后,由SAM(S-腺苷甲硫氨酸)自由基甲基转移酶(radical SAM methyltransferase,RSMT)PhpK在膦酸基团上引入一个甲基,形成第二个碳磷键,并由去乙酰基酶去除氨基上的乙酰基保护,得到最终产物双丙氨膦(3)。
2.1 亚膦酸中间体(含C-P-H结构单元)的形成
与其它膦酸类化合物的合成相同,双丙氨膦的生物合成也开始于磷酸烯醇式丙酮酸变位酶(Ppm)催化的重排反应,这种酶催化磷酸烯醇式丙酮酸(4, PEP)重排生成膦酸丙酮酸(5, P-pyr)。由于化合物4中P-O键的断裂能高于化合物5中的C-P键(约17~24 kcal/mol)[21-22],该反应的平衡常数Keq只有约2×10-4[23]。因此,在生物体内该反应通常与其它生化反应进行耦联,以此来推进反应向生成化合物5的方向进行[23],如脱羧、转氨和羟醛缩合反应等。PEP变位酶是一种重要的催化C-P键形成的酶,其催化的反应是许多膦酸类化合物形成的基础,因此,编码PEP变位酶的ppm基因被用于通过基因序列比对来发现新的膦酸化合物产生菌株。在PEP重排生成化合物5之后,化合物5在磷酸丙酮酸脱羧酶(Ppd)的作用下脱羧生成化合物6。磷酸丙酮酸脱羧酶(Ppd)首次由Seto等[24]从双丙氨膦的产生菌株吸水链霉菌ATCC 21705中分离得到,该酶在催化反应时需要镁离子和硫胺素焦磷酸(TPP)的参与。此后,化合物6在phpC编码的NAD(P)H依赖的氧化还原酶的作用下生成羟乙基膦酸(7, HEP)。羟乙基膦酸不仅是PT及其多肽衍生物等合成的重要中间体,在磷霉素等许多膦酸化合物的生物合成中也至关重要[25]。phpD编码的2-HEP双加氧酶(HEPD)在NADPH或NADH存在的情况下催化HEP的2、3位C-C键断裂形成羟甲基膦酸(8,HMP)[26],HEPD是一种非血红素铁依赖的双加氧酶,在催化C-C键断裂的过程中可能涉及到超氧化铁和氢氧化铁中间体[27]。HMP脱氢酶PhpE、乙醛脱氢酶PhpJ催化后续的两步氧化反应将羟甲基转化为羧基,形成膦甲酸(10, PF)。PF生成后,在PhpF的作用下,CTP(胞嘧啶核苷三磷酸)的β-和γ-位的两个磷酸基团以焦磷酸的形式被PF置换,生成化合物11(CMP-5'-PF)[26]。基因敲除实验显示,吸水链霉菌ATCC 21705中的bcpE基因和羧基膦酸丙酮酸(CPEP)形成有关,对应产绿色链霉菌中与之同源的phpH基因[28]。但PhpH与烯醇酶家族蛋白同源,该家族的蛋白不具备直接催化CMP-PF向CPEP转化的能力[29];而基因簇中的phpG基因编码一种与磷酸甘油酸变位酶同源的蛋白[18],这表明CPEP可能在PhpG和PhpH的共同作用下通过一个两步反应形成。紧接着在CPEP变位酶PhpI的作用下CPEP发生重排反应,产生了亚膦酸中间体亚膦酸丙酮酸(14, PPA),再经一系列羟化、脱羧和转氨等反应,得到双丙氨膦核心骨架草铵膦的前体去甲基草铵膦(18, DMPT)。
2.2 非核糖体三肽的组装
对吸水链霉菌ATCC21705的突变株进行发酵时,在发酵液中发现了乙酰化修饰产物的累积,其中19 AcDMPT是主要的乙酰化中间体[30-31]。随后,在吸水链霉菌ATCC21705中发现了表达N-乙酰基转移酶的bar基因[32],在产绿色链霉菌DSM 40736发现了同样具有此功能的pat基因[33]。对缺失bar基因的吸水链霉菌ATCC 21705进行发酵检测时,发现了化合物18 DMPT的累积,进一步证实了乙酰化修饰是进入三肽合成前的必要步骤[34]。bar/pat基因编码的乙酰基转移酶负责对草铵膦的氨基进行乙酰化修饰,是草铵膦产生菌株进行自我保护的一种机制,可以避免草铵膦对产生菌株自身谷氨酰胺合成酶产生毒性,利用这一特性,bar/pat基因也被用于开发抗草铵膦除草剂的农作物。
乙酰化修饰完成后,化合物19进行肽链的延伸生成三肽中间体20,这一过程是由PhsA/PhsB/PhsC 3个非核糖体肽合成酶催化完成的。NRPS催化非核糖体肽(nonribosomal peptide, NRP)合成时采用模块化的方式,每个模块负责一个氨基的活化,并将其装配到不断延伸的肽链上。其中每个模块包含3个重要的结构域:腺苷化结构域(adenylation domain, A)、硫醇化结构域(thiolation domain, T)、缩合结构域(condensation domain, C)。腺苷化结构域负责氨基酸的选择、活化,并将其装载到硫醇化结构域上。硫醇化结构域又称肽基载体蛋白(PCP)结构域,它带有一个4'-磷酸泛酰巯基乙胺(Ppant)辅基,可以与氨基酸上的羧基结合,并将氨基酸传递到缩合结构域上。缩合结构域负责肽键的形成,在这里新的氨基酸不断地被添加到肽链上[35]。参与三肽中间体20合成的3個非核糖体肽合成酶PhsABC中,PhsA可以识别19(AcDMPT)而不是18(DMPT)作为底物,这是一个很有趣的现象,因为NRPSs通常会选择含有未修饰的α-氨基酸来作为其底物[36]。PhsA对N-乙酰化修饰底物的偏好源自其A-结构域的特殊变化,其中一个活性位点上负责结合α-氨基的天冬氨酸被缬氨酸取代,这种变化只在A-结构域选择非α-氨基酸时出现[37]。PhsB和PhsC各结合了一个丙氨酸,在3个蛋白的共同作用下完成肽链延伸,生成AcDMPT-Ala-Ala三肽。
2.3 次膦酸(含C-P-C结构单元)的形成
完成三肽的合成之后,由PhpK在磷原子上进行甲基化修饰,生成第二个C-P键,形成次膦酸的结构,生成21(乙酰双丙氨膦)。随后,由乙酰基转移酶催化化合物21脱去氮原子上乙酰基,生成最终产物双丙氨膦3,吸水链霉菌中的bah基因、产绿色链霉菌中的dea基因与此过程相关,但尚未得到体外实验验证[20]。PhpK催化的甲基化反应是目前天然产物的生物合成中发现的唯一1例在磷原子上进行甲基化修饰的酶反应[38],PhpK的作用机制尚未明确,但是已有的证据表明,PhpK在SAM的存在下催化甲基从甲基钴胺素转移到AcDMPT上[39]。该反应与磷霉素合成途径中的Fom3催化的反应类似[40],说明PhpK可能是一种B类SAM自由基甲基转移酶(RSMT)。这类酶属于SAM自由基酶(radical SAM enzymes,RS)超家族,该家族酶的一级结构中含有四铁四硫簇[4Fe-4S]的结合位点CXXCXXXC(C:半胱氨酸;X:其它氨基酸)序列。根据B类RSMT的特点,推测PhpK的作用机制为,首先由四铁四硫簇上未与半胱氨酸残基结合的铁原子通过还原切割SAM分子形成5'-dAdo自由基,然后由5'-dAdo自由基拔取化合物20中磷原子上的氢,从而形成底物自由基。底物自由基进攻甲钴胺,拔取甲钴胺上的甲基,完成甲基化修饰的过程,生成化合物21。随后,另外一分子SAM参与到反应中,将甲基传递给钴胺素,形成新的甲钴胺。但是这种机制中尚有存疑:底物-酶-铁硫簇复合物-SAM复合物中,未结合半胱氨酸的特殊铁原子的还原电势低于SAM分子上的硫原子,因此要完成上述的还原切割,需要跨过一个约330 mV的能量壁垒[38]。近年来, Broderick等[41]利用快速冷冻淬灭(rapid freeze-quench, rfq)电子顺磁共振(electron paramagnetic resonance, EPR)和电子核双共振(electron nuclear double-resonance, ENDOR)技术对RS家族中丙酮酸酯酶激酶PFL-AE的研究中捕捉到了一种有机金属中间体,命名为Ω中间体。SAM的C5'-S键被切割,并且SAM的腺苷部分通过C-Fe键结合到铁硫簇上未结合半胱氨酸的铁原子上,形成了该中间体。Ω中间体的发现引发了对于RS作用机制的新的思考:Ω中间体与推测的5'-dAdo自由基的形成存在怎样的关系?这种中间体仅存于PFL-AE的作用过程中还是广泛的参与到RS家族酶的作用过程?
3 草铵膦的降解
草铵膦作为除草剂使用时,在环境中的残留较少,但是对于其在环境中降解的机制,目前尚未有明确的研究。磷元素作为所有已知生命形式的基本元素,是核酸、碳水化合物和磷脂等的组成成分,许多微生物在缺乏磷酸盐作为磷源的情况下可以分解膦酸盐来获取磷元素。在下述的膦酸化合物的降解途径中,或许可以得到关于草铵膦降解机制的启示。微生物分解利用膦酸化合物的方式大致可分为3类:水解酶途径、氧化酶裂解途径和自由基酶裂解途径。
3.1 水解酶途径
水解酶主要有3种。
(1)膦乙醛水解酶(图5a. i) 蜡样芽胞杆菌Bacillus cereus表现出降解22(氨乙基膦酸,2-AEP)的能力[42],其细胞裂解液可以分解化学合成的23(膦乙醛),表明膦乙醛是氨乙基膦酸分解代谢的中间体。Wanner等从鼠伤寒沙门菌Salmonella typhimurium LT2[43]和产气肠杆菌Enterobacter aerogenes[44]中成功克隆到氨乙基膦酸代谢相关的基因,并且成功鉴定出编码转氨酶和膦乙醛水解酶的基因分别为phnW和phnX。序列分析表明,PhnX属于卤酸脱卤酶(HAD)超家族,该家族包括2-卤代烷酸脱卤酶、环氧化物水解酶和磷酸酶[45-47]。
(2)膦酸酯酶(图5a. ii)[46] 22(氨乙基膦酸)还可以通过一个3步反应被降解:转氨、氧化、C-P键水解。化合物22通过转氨、氧化形成的24膦酸酯可以被荧光假单胞菌Pseudomonas fluorescens sp. 23F作为磷源降解利用[48],后经证实,催化膦酸酯降解的酶是phnA编码的膦酸酯水解酶[49]。在苜蓿中华根瘤菌Sinorhizobium meliloti sp. 1021中发现的降解化合物22的基因簇phnWYA进一步证实了这一点,其中phnW/phnY分别编码转氨酶和氧化酶[50]。
(3)膦酸丙酮酸水解酶 这种途径最早在伯克霍尔德菌Burkholderia cepacia sp. Pal6中被发现[51],该菌株可以通过一个两步反应降解25膦酸丙氨酸(图5a. ⅲ):首先在转氨酶的作用下将化合物25转化为膦酸丙酮酸水解酶(PPH)的底物26膦酸丙酮酸,然后在PPH的作用下分解生成27丙酮酸和磷酸,H218O的参与反应时生成18O标记的磷酸,证实PPH催化的反应是一种水解反应[52]。PPH对底物26具有高度专一性,并且其活性依赖Co2+、Ni2+和Mg2+等金属离子的激活[52]。
序列分析发现,在贪噬菌Variovorax sp. Pal2中纯化到的另外的PPH(PalA)与PEP变位酶具有较高的序列同源性[53-54],因此PPH催化C-P键断裂的机制可能与PEP变位酶催化C-P键形成的逆反应类似。
3.2 氧化酶途径
2-AEP是自然界中含量最丰富的膦酸化合物,在对海洋微生物的功能和基因组学解析中發现了2-AEP的新途径:通过phnY和phnZ两个基因的回补,可以使phn基因簇缺失的突变株以化合物22为磷源存活[55]。氨基酸序列分析表明,PhnY是一种α-酮戊二酸(α-KG)依赖的双加氧酶,PhnZ则是金属依赖的膦酸水解酶[56-57],二者可以通过两步氧化反应裂解化合物22中的C-P键,生成α-氨基乙酸和磷酸[61](图5b)。
3.3 自由基酶(C-P裂解酶)途径
相比于其它两类途径,自由基酶途径对底物的宽泛性较高,可以催化膦酸酯、2-氨乙基膦酸、苯基膦酸等的降解,图5c中列举了一系列可以通过该途径进行降解的膦酸化合物[20]。这种途径还可以降解28(二甲基次膦酸)和29(二乙基次膦酸)等含有两个C-P键的次膦酸化合物;同时相比于其它途径,这种途径也是在微生物中分布最为广泛的,在一些革兰阳性菌如节杆菌Arthrobacter sp. GLP-1、巨大芽胞杆菌Baeillusmegatherium,以及许多革兰阴性菌如大肠埃希菌Escherichia coli、假单胞菌、根瘤菌Rhizobium sp.、放射土壤杆菌Agrobacterium radiobacter、克雷伯杆菌Klebsiella sp.等中均有分布[59]。C-P键裂解酶的活性最早在1963年发现于大肠埃希菌中[60],但是直到近年来其催化C-P键裂解的机制才被揭示。C-P键裂解酶途径涉及到phnCDEFGHIJKLMNOP共14个基因组成的基因簇。PhnCDE是一组ABC型的转运蛋白,其中PhnD是结合蛋白,充当“守门员”的功能,可以特异性地识别膦酸基团[61];PhnE是跨膜区;PhnC是驱动膦酸化合物跨细胞膜转入细胞的ATP依赖蛋白[62]。在进入细胞后,以22(氨乙基膦酸)为例,C-P裂解酶途径降解膦酸的途径如图4d。首先PhnO在乙酰辅酶A的辅助下进行氨乙酰化修饰,该步骤非必要步骤,在R基团为甲基、乙基等烷基的化合物降解途径中并不发生。在PhnGHL的辅助下,PhnI催化对膦酸连接到ATP分子的核糖上,生成31(核糖-1-膦酸酯-5-三磷酸),PhnM则催化三磷酸基团裂解去除焦磷酸[63]。随后PhnJ催化C-P键断裂,这是该途径的核心步骤,PhnJ是一个四铁四硫簇[4Fe-4S]和SAM依赖的蛋白[63-64], 催化C-P键裂解的机制与SAM自由基酶作用机制一致:首先是四铁四硫簇对SAM分子还原切割形成5'-dAdo自由基,自由基传递到PhnJ活性位点的氨基酸残基上之后蛋白与膦酸基团的磷原子结合,再通过自由基的传递使C-P键断裂形成碳自由基;碳自由基拔取β-C上羟基的氢原子,羟基氧原子与磷原子结合形成33(环状α-D-核糖基磷酸酯)。PhnP是金属β-内酰胺酶超家族的成员之一[65],它可以对33的环磷酸进行区域特异性水解,得到细胞可利用的代谢物34 α-D-核糖基-1, 5二磷酸,然后由PhnN进一步磷酸化修饰后,生成35(5'-磷酸-α-D-核糖基-二磷酸, PRPP),进入细胞的代谢途径中。
草铵膦有着特殊的C-P-C键结构单元,含有两个碳磷键,其降解途径目前并不十分明确。正如“3.3”部分所说,自由基酶机制(C-P裂解酶)催化的C-P键裂解可以将次膦酸作为底物。因此,草铵膦的降解很可能选择这一底物宽泛性较强的途径,区别在于在其中一个C-P键断裂后膦酸基团并非连接在核糖上进入生物代谢,而是从核糖上脱落进入下一个C-P键裂解反应。
4 讨论与展望
草铵膦生物合成途径的解析尚存在一些谜点,其中PhpK催化的P-甲基化反应广受关注。PhpK催化的P-甲基化反应极其特殊,解析其催化反应的机制不仅是解析草铵膦生物合成途径的关键,同时也具有重要的生物化学意义,是打开次膦酸化合物生物化学宝库的金钥匙。同时,C-P-C结构单元生物合成途径中PhpK催化的P-甲基化反应与C-P裂解酶途径中催化C-P键断裂的反应都涉及到SAM自由基酶机制,对PhpK反应机制的解析可以帮助我们理解C-P键断裂的机制,可能会为次膦酸化合物的生物降解研究提供思路。目前关于草铵膦的生物降解机制尚不明确,解析草铵膦的生物降解方式对于草铵膦的合理施用,减小除草剂施用对环境的危害将起到巨大的推动作用。
参 考 文 献
Troyer J R. In the beginning: The multiple discovery of the first hormone herbicides[J]. Weed Sci, 2001, 49(2): 290-297.
杨益军, 张波. 2020年全球(中国)草铵膦市场状况分析及预测[J]. 世界农药, 2020, 42 (3): 20-30.
张一宾. 近年来全球草铵膦的市场及发展趋向[J]. 农药, 2016, 55(5): 313-315, 323.
Duke S. The history and current status of glyphosate[J]. Pest Manag Sci, 2018, 74(5): 1027-1034.
Belz R G, Duke S O. Herbicides and plant hormesis[J]. Pest Manag Sci, 2014, 70(5): 698-707.
Samac D A, Foster-Hartnett D. Effect of glyphosate application on foliar diseases in glyphosate-tolerant alfalfa[J]. Plant Dis, 2015, 96(8): 1104-1110.
Bayer E, Gugel K H, H?gele K, et al. Stoffwechselprodukte von mikroorganismen. 98. Mitteilung. phosphinothricin und Phosphinothricyl-Alanyl-Alanin[J]. Helv Chim Acta, 1972, 55(1): 224-239.
Kondo Y. Studies on a new antibiotic SF-1293. I. Isolation and physico-chemical and biological characterization of SF-1293 sub-stance[J]. Rep Mjiska, 1973, 13: 34-41.
Omura S, Hinotozawa K, Imamura N, et al. The structure of phosalacine, a new herbicidal antibiotic containing phosphinothricin[J]. J Antibiot, 1984, 37(8): 939-940.
Omura S, Murata M, Hanaki H, et al. Phosalacine, a new herbicidal antibiotic containing phosphinothricin. Fermentation, isolation, biological activity and mechanism of action[J]. J Antibiot, 1984, 37(8): 829-835.
Kato H, Nagayama K, Abe H, et al. Isolation, structure and biological activity of trialaphos[J]. Agric and Biol Chem, 1991, 55(4): 1133-1134.
Jablonkai I. Molecular mechanism of action of herbicides[M]. InTech, 2011.
Frieg B, G?rg B, Gohlke H, et al. Glutamine synthetase as a central element in hepatic glutamine and ammonia metabolism: Novel aspects[J]. Biol Chem, 2021, 402(9): 1063-1072.
Unno H, Uchida T, Sugawara H, et al. Atomic structure of plant glutamine synthetase[J]. J Biol Chem, 2006, 281(39): 29287-29296.
Block M, Botterman J, Vandewiele M, et al. Engineering herbicide resistance in plants by expression of a detoxifying enzyme[J]. Embo J, 1987, 6(9): 2513-2518.
陳国强, 吕武华, 邓畅, 等. 草铵膦合成技术的研究进展[J]. 化学世界, 2021, 62(2): 65-70.
Schwartz D, Berger S, Heinzelmann E, et al. Biosynthetic Gene Cluster of the Herbicide Phosphinothricin Tripeptide from Streptomyces viridochromogenes Tü494[J]. Appl Environ Microbiol, 2004, 70(12): 7093-7102.
Blodgett J a V, Zhang J K, Metcalf W W. Molecular cloning, sequence analysis, and heterologous expression of the phosphinothricin tripeptide biosynthetic gene cluster from Streptomyces viridochromogenes DSM 40736[J]. Antimicrob Agents Chemother, 2005, 49(1): 230-240.
Joshua, Blodgett, Jun, et al. Conserved biosynthetic pathways for phosalacine, bialaphos and newly discovered phosphonic acid natural products[J]. J Antibiot, 2016, 69(1): 15-25.
Horsman G P, Zechel D L. Phosphonate biochemistry[J]. Chem Rev, 2017, 117(8): 5704-5783.
Hemelsoet K, Van Durme F, Van Speybroeck V, et al. Bond dissociation energies of organophosphorus compounds: An assessment of contemporary ab initio procedures[J]. J Phys Chem, 2010, 114(8): 2864-2873.
Monique N, Al-Abdul-Wahid M, Krystal F, et al. Understanding the mechanism of action of triazine-phosphonate derivatives as flame retardants for cotton fabric[J]. Molecules, 2015, 20(6): 11236-11256.
Kim J, Dunaway-Mariano D. Phosphoenolpyruvate mutase catalysis of phosphoryl transfer in phosphoenolpyruvate: kinetics and mechanism of Phosphorus-Carbon bond formation[J]. Biochemistry, 1996, 35(14): 4628-4635.
Hidaka T, Mori M, Imai S, et al. Studies on the biosynthesis of bialaphos (SF-1293). 9. Biochemical mechanism of C-P bond formation in bialaphos: discovery of phosphoenolpyruvate phosphomutase which catalyzes the formation of phosphonopyruvate from phosphoenolpyruvate[J]. J Antibiot, 1989, 42(3): 491-494.
Blodgett J, Thomas P M, Li G, et al. Unusual transformations in the biosynthesis of the antibiotic phosphinothricin tripeptide[J]. Nat Chem Biol, 2007, 3(8): 480-485.
Cicchillo R M , Zhang H, Blodgett J, et al. An unusual carbon-carbon bond cleavage reaction during phosphinothricin biosynthesis[J]. Nature, 2009, 459(7248): 871-874.
Hirao H, Morokuma K. Ferric superoxide and ferric hydroxide are used in the catalytic mechanism of hydroxyethylphosphonate dioxygenase: A density functional theory investigation[J]. J Am Chem Soc, 2010, 132(50): 17901-17909.
Lee S H, Hidaka T, Nakashita H, et al. The carboxypho-sphonoenolpyruvate synthase-encoding gene from the bialaphos-producing organism Streptomyces hygroscopicus[J]. Gene, 1995, 153(1): 143-144.
Gerlt J A, Babbitt P C, Jacobson M P, et al. Divergent evolution in enolase superfamily: Strategies for assigning Functions[J]. J Biol Chem, 2015, 287(1): 29-34.
Imai S, Seto H, Sasaki T, et al. Studies on the biosynthesis of bialaphos (SF-1293). 6. Production of N-acetyl-demethylphosphinothricin and N-acetylbialaphos by blocked mutants of Streptomyces hygroscopicus SF-1293 and their roles in the biosynthesis of bialaphos[J]. J Antibiot, 1985, 38(5): 687-690.
Hara O, Anzai H, Imai S, et al. The bialaphos biosynthetic genes of Streptomyces hygroscopicus: cloning and analysis of the genes involved in the alanylation step[J]. J Antibiot, 1988, 41(4): 538-547.
Thompson C J, Movva N R, Tizard R, et al. Characterization of the herbicide-resistance gene bar from Streptomyces hygroscopicus[J]. Embo J, 1987, 6(9): 2519-2523.
Strauch E, Wohlleben W, Pühler A. Cloning of a phosphinothricin N-acetyltransferase gene from Streptomyces viridochromogenes Tü494 and its expression in Streptomyces lividans and Escherichia coli[J]. Gene, 1988, 63(1): 65-74.
Kumada Y, Anzai H, Takano E, et al. The bialaphos resistance gene (bar) plays a role in both self-defense and bialaphos biosynthesis in Streptomyces hygroscopicus[J]. J Antibiot, 1988, 41(12): 1838-1845.
Sussmuth R D, Mainz A. Nonribosomal peptide synthesis-principles and prospects[J]. Angew Chem Int Ed Engl, 2017, 56(14): 3770-3821.
Grammel N, Schwartz D, Wohlleben W, et al. Phosphinothricin-tripeptide synthetases from Streptomyces viridochromogenes[J]. Biochemistry, 1998, 37(6): 1596-1603.
Blodgett J A, Zhang J K, Yu X, et al. Conserved biosynthetic pathways for phosalacine, bialaphos and newly discovered phosphonic acid natural products[J]. J Antibiot, 2016, 69(1): 15-25.
Broderick J B, Duffus B R, Duschene K S, et al. Radical S-adenosylmethionine enzymes[J]. Chem Rev, 2014, 114(8): 4229-4317.
Werner W J, Allen K D, Hu K, et al. In vitro phosphinate methylation by PhpK from Kitasatospora phosalacinea[J]. Biochemistry, 2011, 50(42): 8986-8988.
Allen K D, Wang S C. Initial characterization of Fom3 from Streptomyces wedmorensis: The methyltransferase in fosfomycin biosynthesis[J]. Arch Biochem Biophys, 2014, 543: 67-73.
Horitani M, Shisler K, Broderick W E, et al. Radical SAM catalysis via an organometallic intermediate with an Fe-[5-C]-deoxyadenosyl bond[J]. Science, 2016, 352(6287): 822-825.
La Nauze J M, Rosenberg H. The identification of 2-phosphonoacetaldehyde as an intermediate in the degradation of 2-aminoethylphosphonate by Bacillus cereus[J]. Biochimica et Biophysica Acta, 1968, 165(3): 438-447.
Jiang W, Metcalf W W, Lee K S, et al. Molecular cloning, mapping, and regulation of Pho regulon genes for phosphonate breakdown by the phosphonatase pathway of Salmonella typhimurium LT2[J]. J Bacteriol, 1995, 177(22): 6411-6421.
Lee K, Metcalf W, Wanner B. Evidence for two phosphonate degradative pathways in Enterobacter aerogenes[J]. J Bacteriol, 1992, 174(8): 2501-2510.
Baker A S, Ciocci M J, Metcalf W W, et al. Insights into the mechanism of catalysis by the P-C bond-cleaving enzyme phosphonoacetaldehyde hydrolase derived from gene sequence analysis and mutagenesis[J]. Biochemistry, 1998, 37(26): 9305-9315.
Nauze J, Rosenberg H, Shaw D C. The enzymic cleavage of the carbon-phosphorus bond: Purification and properties of phosphonatase[J]. Biochimica et Biophysica Acta, 1970, 212(2): 332-350.
Dumora C, Marche M, Doignon F, et al. First characterization of the phosphonoacetaldehyde hydrolase gene of Pseudomonas aeruginosa[J]. Gene, 1997, 197(1-2): 405-412.
Mcmullan G, Harrington F, Quinn J. Metabolism of phosphonoacetate as the sole carbon and phosphorus source by an environmental bacterial isolate[J]. Appl Environ Microb, 1992, 58(4): 1364-1366.
Kulakova A N, Kulakov L A, Akulenko N V, et al. Structural and functional analysis of the phosphonoacetate hydrolase (phnA) gene region in Pseudomonas fluorescens 23F[J]. J Bacteriol, 2001, 183(11): 3268-3275.
Borisova S A, Christman H D, Metcalf M M, et al. Genetic and biochemical characterization of a pathway for the degradation of 2-aminoethylphosphonate in Sinorhizobium meliloti 1021[J]. J Biol Chem, 2011, 286(25): 22283-22290.
Ternan N G, Quinn J P. In vitro cleavage of the carbon-phosphorus bond of phosphonopyruvate by cell extracts of an environmental Burkholderia cepacia isolate[J]. Biochem Bioph Res Co, 1998, 248(2): 378-381.
Ternan N G, Hamilton J T G, Quinn J P. Initial in vitro characterisation of phosphonopyruvate hydrolase, a novel phosphate starvation-independent, carbon-phosphorus bond cleavage enzyme in Burkholderia cepacia Pal6[J]. Arch Microbiol, 2000, 173(1): 35-41.
Kulakova A N, Wisdom G B, Kulakov L A, et al. The purification and characterization of phosphonopyruvate hydrolase, a novel carbon-phosphorus bond cleavage enzyme from Variovorax sp. Pal2[J]. J Biol Chem, 2003, 278(26): 23426-23431.
Kulakova A N, Kulakov L A, Villarreal-Chiu J F, et al. Expression of the phosphonoalanine-degradative gene cluster from Variovorax sp. Pal2 is induced by growth on phosphonoalanine and phosphonopyruvate[J]. Fems Microbiol Lett, 2010(1): 100-106.
Martinez A, Tyson G W, Delong E F. Widespread known and novel phosphonate utilization pathways in marine bacteria revealed by functional screening and metagenomic analyses[J]. Environ Microbiol, 2010, 12(1): 222-238.
Watanabe M, Sumida N, Murakami S, et al. A phosphonate-induced gene which promotes penicillium-mediated bioconversion of cis-propenylphosphonic acid to fosfomycin[J]. Appl Environ Microb, 1999, 65(3): 1036-1044.
White A K, Metcalf W W. Isolation and biochemical characterization of hypophosphite/2-oxoglutarate dioxygenase: A novel phosphorus-oxidizing enzyme from Pseudomonas stutzeri WM88[J]. J Biol Chem, 2002, 277(41): 38262-38271.
Mcsorley F R, Wyatt P B, Martinez A, et al. PhnY and PhnZ comprise a new oxidative pathway for enzymatic cleavage of a carbon-phosphorus bond[J]. J Am Chem Soc, 2012, 134(20): 8364-8367.
White A K, Metcalf W W. Microbial metabolism of reduced phosphorus compounds[J]. Annu Rev Microbiol, 2007, 61: 379-400.
Zeleznick L D, Myers T C, Titchener E B. Growth of Escherichia coli on methyl- and ethylphosphonic acids[J]. Biochim Biophys Acta, 1963, 78(3): 546-547.
Dyhrman S, Chappell P, Haley S, et al. Phosphonate utilization by the globally important marine diazotroph Trichodesmium[J]. Nature, 2006, 439(7072): 68-71.
Iqbal S, Parker G, Davidson H, et al. Reversible phase variation in the phnE gene, which is required for phosphonate metabolism in Escherichia coli K-12[J]. J Bacteriol, 2004, 186(18): 6118-6123.
Kamat S S, Williams H J, Raushel F M. Intermediates in the transformation of phosphonates to phosphate by bacteria[J]. Nature, 2011, 480(7378): 570-573.
Seweryn P, Van L B, Kjeldgaard M, et al. Structural insights into the bacterial carbon-phosphorus lyase machinery[J]. Nature, 2015, 525(7567): 68-72.
Podzelinska K, He S M, Wathier M, et al. Structure of PhnP, a phosphodiesterase of the carbon-phosphorus lyase pathway for phosphonate degradation[J]. J Biol Chem, 2009, 284(25): 17216-17226.
