固相支撑液液萃取结合LC-MS/MS 快速测定生乳中32 种农药残留
2023-08-25黄佳佳曾上敏张静文
邓 龙,周 思 ,黄佳佳,曾上敏,张静文
(1.广东食品药品职业学院,广东广州 510520;2.广州市疾病预防控制中心,广东广州 510440)
乳及乳制品营养丰富,是人们生活中的重要食品之一。生乳作为配方乳粉、液体乳及其他乳制品的主要原料,其质量安全问题向来是消费者和行业关注的焦点。研究表明,包括生乳在内的动物源性食品不仅受到兽药残留、金属元素等危害因素的影响,还面临着农药残留的污染等问题[1]。我国作为农业大国,农药用量大、利用率低等问题普遍。环境中的农药残留通过食物、饮水、杀虫、环境接触等途径进入动物体内,易在脂肪含量较高的乳类等产品中蓄积,通过食物链持续影响人类健康[2-3]。对此,食品法典委员会、欧盟等制定了严格的限量标准以降低其对健康造成的风险[4-5]。我国GB 2763-2021 规定了生乳中125 种农药残留的最大限量,涉及检验方法25 项[6]。存在操作复杂、项目不全、检出限高于限量值等问题,限量标准配套方法尚不完善[7-8]。文献报道动物源性食品中农残检测多为肉及其制品、水产品、鸡蛋等[9-11],有关生乳的检测项目少、种类单一[10,12],难以满足生乳中农药多残留快速筛查的要求[12]。因此,建立生乳中农药多残留快速检测方法对保障乳制品食品安全具有现实意义。
生乳中农药多残留检测面临两大挑战:a. 目标物种类多,性质差异大,用同一种方法难以获得好的回收率;b. 含量水平低,基质效应强,前处理复杂,生乳不易保存、时效性要求高[13]。传统的液液萃取、固相萃取处理步骤多、操作繁琐[14-15],QuEChERS 影响因素多、方法摸索耗时长[16-17]、凝胶色谱需专用设备[18]等,简单、高效的样品前处理方法有待进一步研究[19]。固相支撑液液萃取(supported liquid extraction,SLE)是1997 年Johnson 团队提出的一种新型的样品前处理技术[20]。以均匀多孔材料为支撑介质,吸附水性样品形成理想的液膜萃取界面,加入与水不相溶的洗脱溶剂后,溶剂与样本之间形成连续的浓度差,从而实现目标物的高效萃取,萃取流程见图1。SLE 在生物样本中药物、环境污染物、烟草及其代谢物的检测中应用广泛[21-25]。SLE 本质是液液萃取,但克服了传统液液萃取效率低、易乳化等缺点,且操作十分简单,只需上样和洗脱两个步骤。在水系样本有机物萃取方面优势明显,可用于生乳中农药多残留样品的前处理,在农药残留方面的应用暂无相关报道。
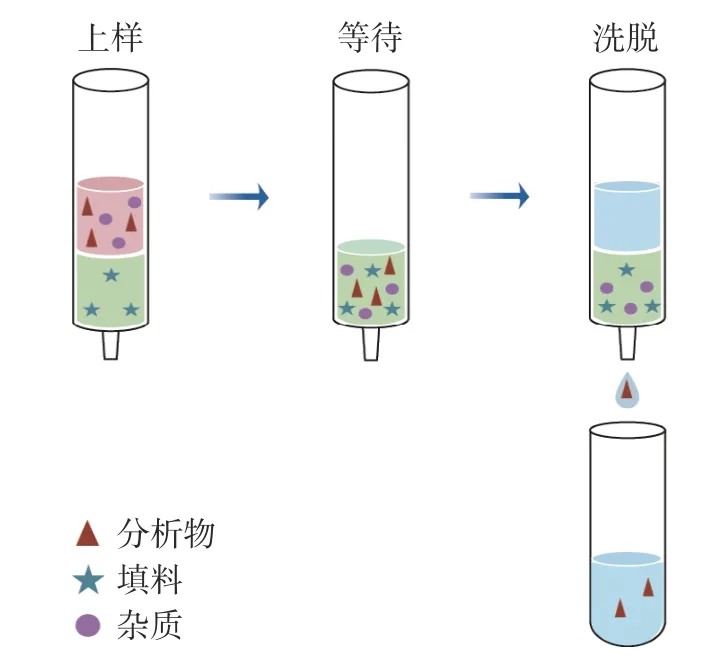
图1 SLE 实验流程示意图Fig.1 Schematic diagram of the experimental procedures of SLE
本研究以农业农村部《禁限用农药名录》中32 种高风险禁限用农药残留为目标物,采用固相支撑液液萃取进行样品前处理,结合液相色谱串联质谱在多残留分析中灵敏度高、速度快、抗干扰能力强的优势[10-12],建立生乳中32 种农药残留的超高效液相色谱-串联质谱检测方法,为动物源性样品中农药多残留的检测提供新思路。
1 材料与方法
1.1 材料与仪器
32 种农药残留标准品 100 mg/L,农业部环境保护科研监测所;乙腈、甲酸铵、甲酸 LC-MS 级,德国Merck 公司;二氯甲烷、正己烷、乙酸乙酯 HPLC级,德国Merck 公司;去离子水 18.0 MΩ·cm,由Milli-Q 去离子水发生器制备;生乳样品 采自本地乳品生产企业及鲜乳配送点,4 ℃冷藏保存。
ACQUITY Xevo TQ-S 超高效液相色谱-串联质谱仪、Wat200609 固相萃取装置 美国Waters 公司;IKA MS3 漩涡混合器 德国IKA 公司;5810R高速冷冻离心机 德国Eppendorf 公司;N-EVAP112氮吹浓缩仪 美国Organomation 公司;Milli-Q 去离子水发生器 美国Millipore 公司;SLE 萃取柱 3 mL美国Agilent 公司;Mini-UniPrep 聚四氟乙烯非针式过滤器 0.2 μm 英国Whatman 公司。
1.2 实验方法
1.2.1 标准溶液配制 准确吸取0.1 mL 单标溶液于10 mL 容量瓶,乙腈定容至刻度,配制成1 mg/L混合标准溶液,用生乳空白基质溶液稀释为0.2、1、2、5、10、20、50 μg/L 系列标准工作溶液。
1.2.2 样品前处理 准确称取1 g(精确至0.01 g)混匀的生乳样品,注入装有1.5 mL 乙腈的离心管,涡旋混匀,4000 r/min 离心5 min,取上清液。样品沉淀加入0.5 mL 水冲洗,合并清液,转移至SLE 小柱,等待5 min。用4.5 mL 二氯甲烷洗脱2 次,待无液滴落下后抽真空5 s,洗脱液氮吹至近干,用20%乙腈甲酸(0.1%)水溶液定容至0.5 mL,非针式过滤器过滤。
1.2.3 色谱条件 色谱柱:Waters CORTECS C18柱(100 mm×2.1 mm,2.7 μm);流动相:A 为0.1%甲酸溶液(含5 mmol/L 甲酸铵),B 为0.1%甲酸乙腈(含5 mmol/L 甲酸铵),梯度洗脱程序:0~2 min,20% B;2~13 min,B 升至100%;13~15 min,保持100% B;15~16 min,B 降至20%;16~18 min,保持20% B;进样体积:5 μL;流速:0.30 mL/min;柱温:30 ℃。
1.2.4 质谱条件 离子源:电喷雾离子源(ESI),氟虫腈为负离子模式,其余31 种为正离子模式;监测方式:多反应监测(MRM)模式;毛细管电压:3.0 kV;离子源温度:150 ℃;脱溶剂气温度:450 ℃;各化合物参考保留时间、监测离子、锥孔电压、碰撞电压等参数见表1。
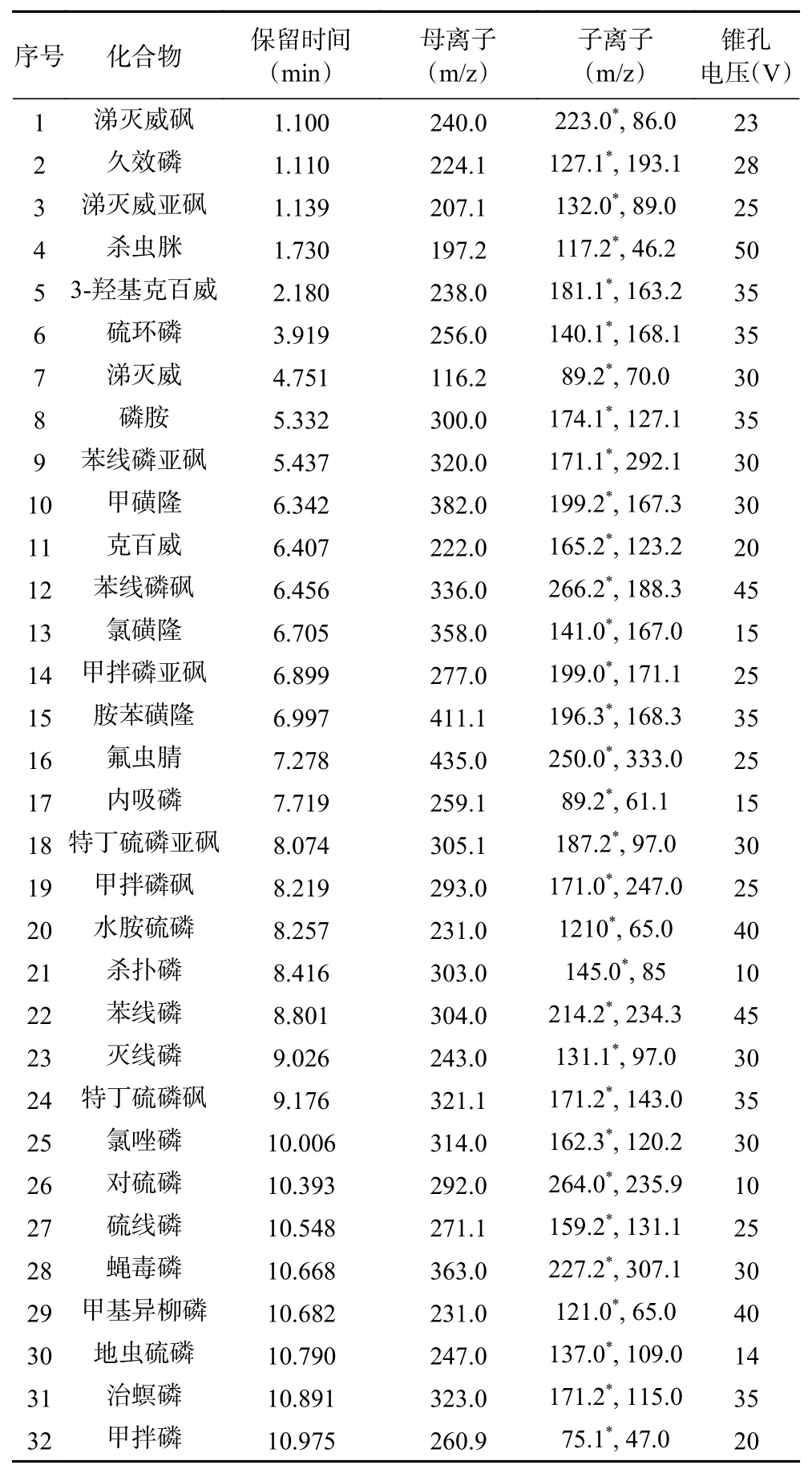
表1 32 种目标物的保留时间和质谱参数Table 1 Retention times and mass parameters of target compounds
1.3 数据处理
采用基质匹配校准曲线外标法定量,通过标准加入法和控制变量法优化实验条件。应用仪器配套Masslynx 工作站进行数据采集与标准曲线绘制,导出数据使用Origin 2017 进行数据分析与作图。前处理和分析条件优化中实验重复测试3 次取均值,回收率与精密度实验重复测试6 次计算相对标准偏差。
2 结果与分析
2.1 预处理条件优化
生乳含有大量的蛋白、脂肪和磷脂等物质,易残留在液质系统中,改变分析物峰型,增加色谱柱压力,影响离子化效率,上机测试前应尽量去除[26],固相支撑液液萃取以改性多孔材料为支撑,表面活性低、比表面积大,可形成微孔液膜实现目标物的高效萃取。同时,保留样本中的磷脂、蛋白等干扰物,有效减少基质干扰[27]。SLE 只有上样和洗脱两个步骤,上柱样本状态及洗脱液的选取是影响实验的关键因素[25]。上样前对样本进行稀释、调酸碱度、沉淀蛋白等预处理可以获得更好的萃取与净化效果[23]。生乳含有大量蛋白直接上柱易造成柱子堵塞影响柱效,采用有机试剂沉淀蛋白后上柱,可大幅降低样品中的蛋白含量,且水溶性有机溶剂的加入可改善部分极性化合物萃取率[28]。实验比较了甲醇、乙腈、0.1%甲酸乙腈三种溶剂的沉淀效果,乙腈沉淀效果优于甲醇,少量酸的加入有利于稳定酸性农药但不利于蛋白沉淀,故选择乙腈为沉淀剂。比较加入0.5、1.0、1.5、2.0 mL 乙腈的沉淀效果,不同体积乙腈沉淀效果见图2,当加入体积达到1.5 mL,离心后样本呈澄清状态,且最终回收率在60%~120%之间,符合GB/T 27404 对回收率的要求,故选择1.5 mL 乙腈进行沉淀。此外,将生乳样品注入乙腈中,可以让样本与溶剂充分接触,沉淀效果更好。
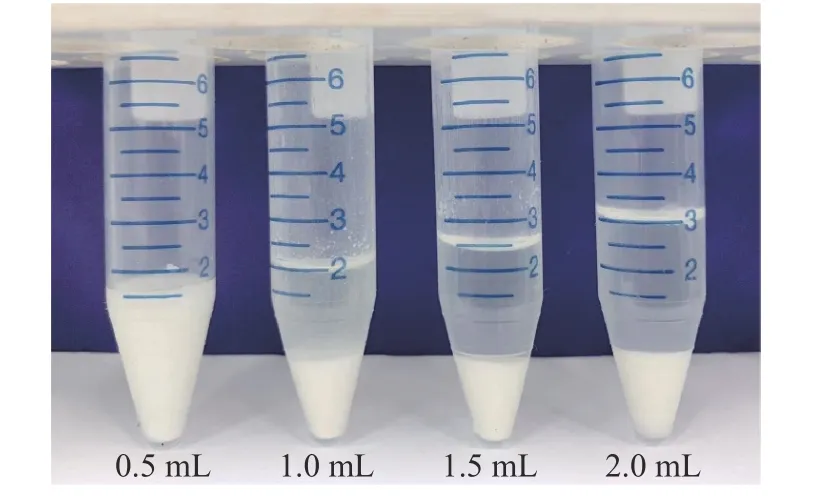
图2 不同乙腈用量对生乳样品的沉淀效果Fig.2 Precipitation effect of different acetonitrile volumes on raw milk samples
2.2 萃取柱条件优化
固相支撑液液萃取的本质是目标物在两相中的分配,32 种目标化合物中含有机磷、氨基甲酸酯、磺酰脲、苯基吡唑等多类化合物,油水分配系数差异大,洗脱溶剂的选择需充分考虑目标物的极性和溶解度。实验考察乙酸乙酯、乙腈、二氯甲烷、正己烷四种常用溶剂的洗脱效果。结果表明,正己烷极性小,对苯线磷、涕灭威、久效磷等水溶性较好的农药洗脱效果不佳。乙腈极性大,与水性样本互溶,容易造成样品穿漏,影响净化效果。乙酸乙酯洗脱甲基异柳磷、蝇毒磷、氟虫腈回收率低于20.0%,二氯甲烷对各目标化合物的回收率均较理想,平均回收率为69.4%~113.6%,故选择二氯甲烷作为洗脱溶剂。进一步优化洗脱溶剂用量,考察溶剂体积分别为柱容量2、3、4 倍时的洗脱效果,加标浓度为10 μg/kg 时总体回收率比较见图3。由图可知,当体积达到9 mL时全部目标物回收率均超过60.0%,平均回收率为89.8%,继续增加溶剂体积总体回收率增幅不明显,基于后续操作及环保考虑不再增加溶剂用量。根据分配定律,同样体积的溶剂多次萃取比单次萃取效率高,比较9 mL×1 和4.5 mL×2 洗脱效果,结果与理论一致,最终选择用4.5 mL 二氯甲烷洗脱2 次。
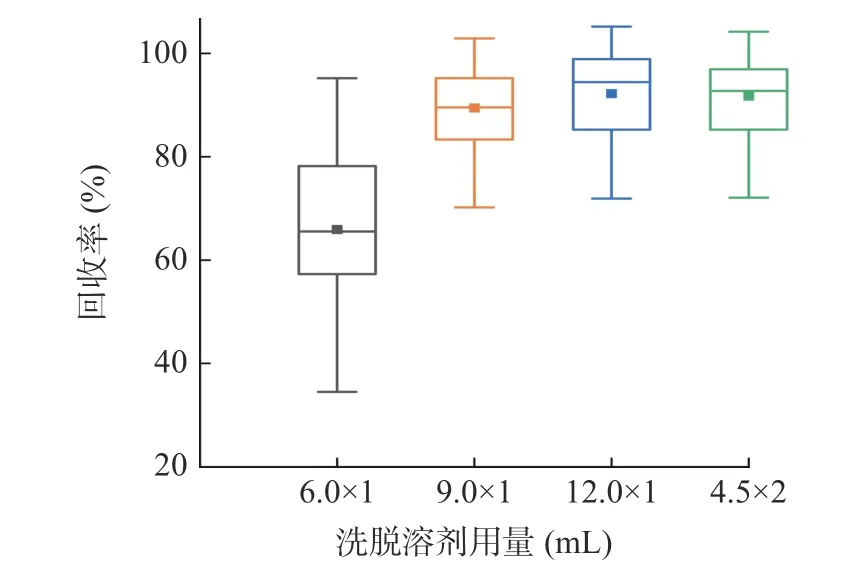
图3 目标化合物回收率与洗脱溶剂用量的关系Fig.3 Relationship between the recovery of target compounds and the amount of elution solvent
2.3 色谱条件优化
液相色谱串联质谱分析农药多残留,流动相多选择水、甲醇或乙腈的流动相体系。实验对比了几种常用流动相的分离效果,水相包括0.1%甲酸、5 mmol/L 甲酸铵、5 mmol/L 甲酸铵(含0.1%甲酸),有机相包括5 mmol/L 甲酸铵甲醇、5 mmol/L 甲酸铵乙腈、5 mmol/L 甲酸铵乙腈(含0.1%甲酸)。实验发现,5 mmol/L 甲酸铵(含0.1%甲酸)-5 mmol/L甲酸铵乙腈(含0.1%甲酸)流动相体系分离效果最佳[17]。乙腈洗作为流动相洗脱力强峰型更加尖锐对称。目标物多为酸性农药,加入少量甲酸有利于[M-H]+离子的生成,保持化合物稳定,减少基质中共萃物的干扰,提高方法的稳定性和重现性。加入甲酸铵有利于[M-H]-离子的生成,增强离子响应强度,同时降低保留时间长的化合物的柱保留,可有效改善色谱峰型[9]。初始流动相水相比例过高久效磷等极性较大的化合物难以保留,易与极性基质一起流出影响测定。调低初始水相比例至80%,保持2 min,有利于极性化合物的分离与测定。
2.4 基质效应评价
基质效应(matrix effect,ME)是指受样品基质影响导致化合物响应值增强或抑制的现象[29]。痕量水平残留检测易受基质效应影响,目前有关动物源性样本对农药残留的基质效应的研究较少。本实验用生乳空白基质标曲与纯溶剂标曲的斜率比值对基质效应大小进行评估。ME 值大于1.2 为基质增强,ME值小于0.8 为基质抑制,ME 值在0.8~1.2 认为基质效应可接受,32 种农残ME 值分布见图4。32 种目标化合物产生基质抑制效应的农残占比34.4%,产生基质增强效应的农残占比9.4%。目标物在生乳中的基质效应不可忽略,实验中采用基质匹配标准曲线减小基质效应,提高测定结果的准确度。
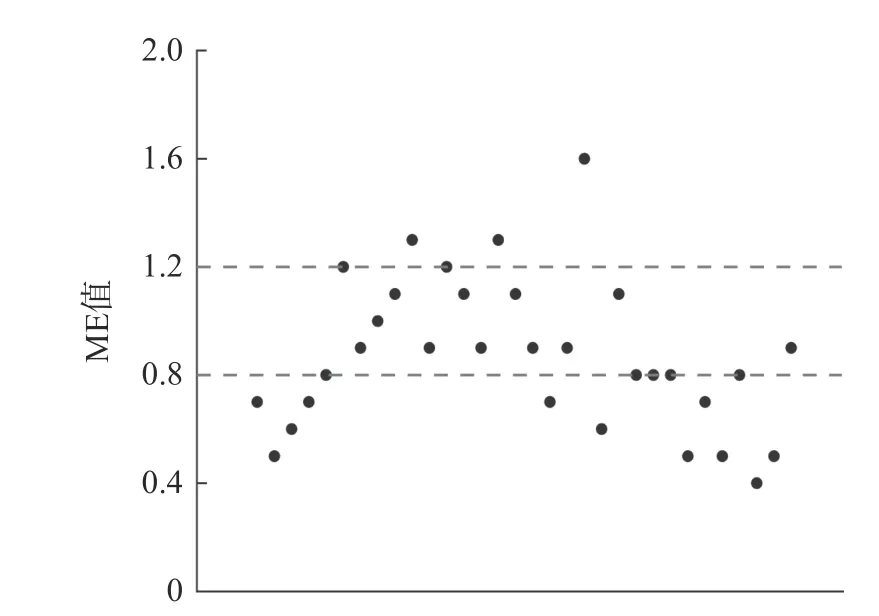
图4 目标化合物ME 值分布图Fig.4 Matrix effect distribution of target compounds
2.5 线性范围和检出限
在优化条件下测试1.2.1 配制的基质匹配标准工作溶液。以目标物峰面积(y)对质量浓度(x,μg/L)进行线性拟合,得到32 种农药残留的线性方程(见表2)。由表可知,32 种目标物在相应浓度范围内线性关系良好,相关系数均大于0.9962。选择低添加水平样品,以3 倍信噪比(S/N=3)计算检出限,以10 倍信噪比(S/N=10)计算定量限,结合样品前处理过程,得到检出限为0.1~2.5 μg/kg,定量限为0.3~7.5 μg/kg。
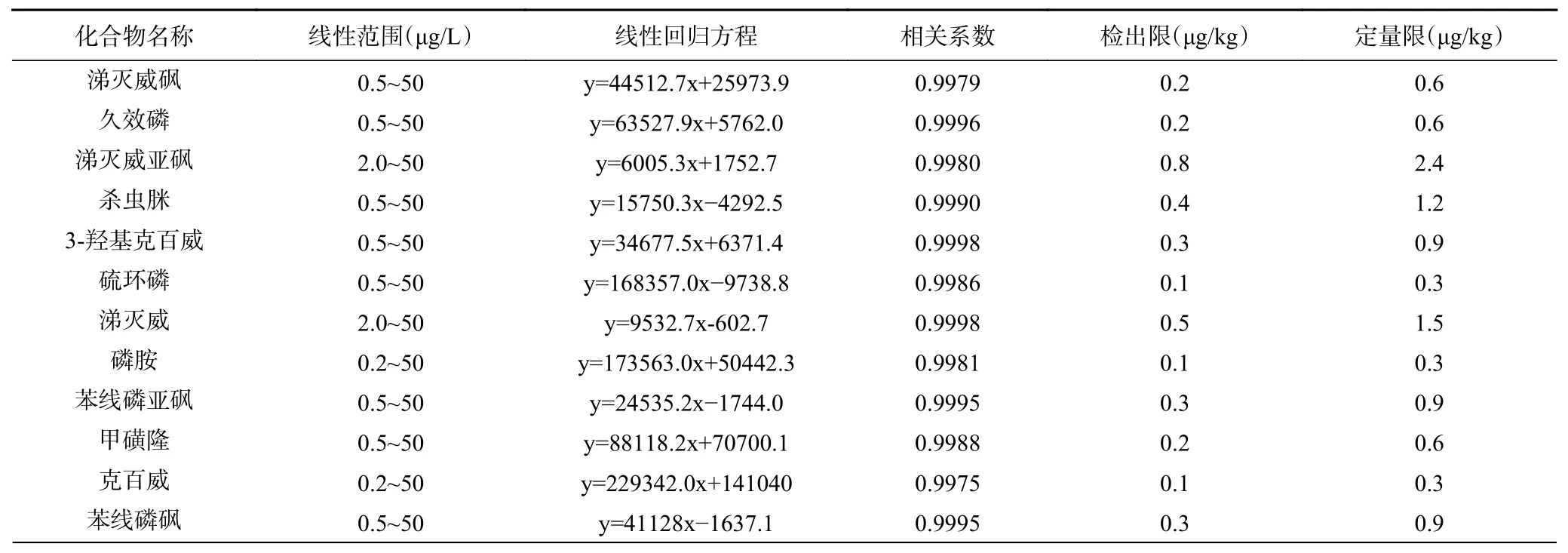
表2 线性范围、线性方程、相关系数、方法检出限和定量限Table 2 Linear equations, linear ranges, correlation coefficients, detection limits and quantitation limits
2.6 回收率与精密度
选取阴性生乳样品,开展低、中、高3 水平加标回收实验,平行测试6 次,结果见表3。32 种目标物平均回收率为69.4%~113.8%,相对标准偏差(RSD,n=6)为1.4%~8.2%,符合GB/T 27404[30]对食品理化检测的质量控制要求,方法具有良好的回收率与精密度。
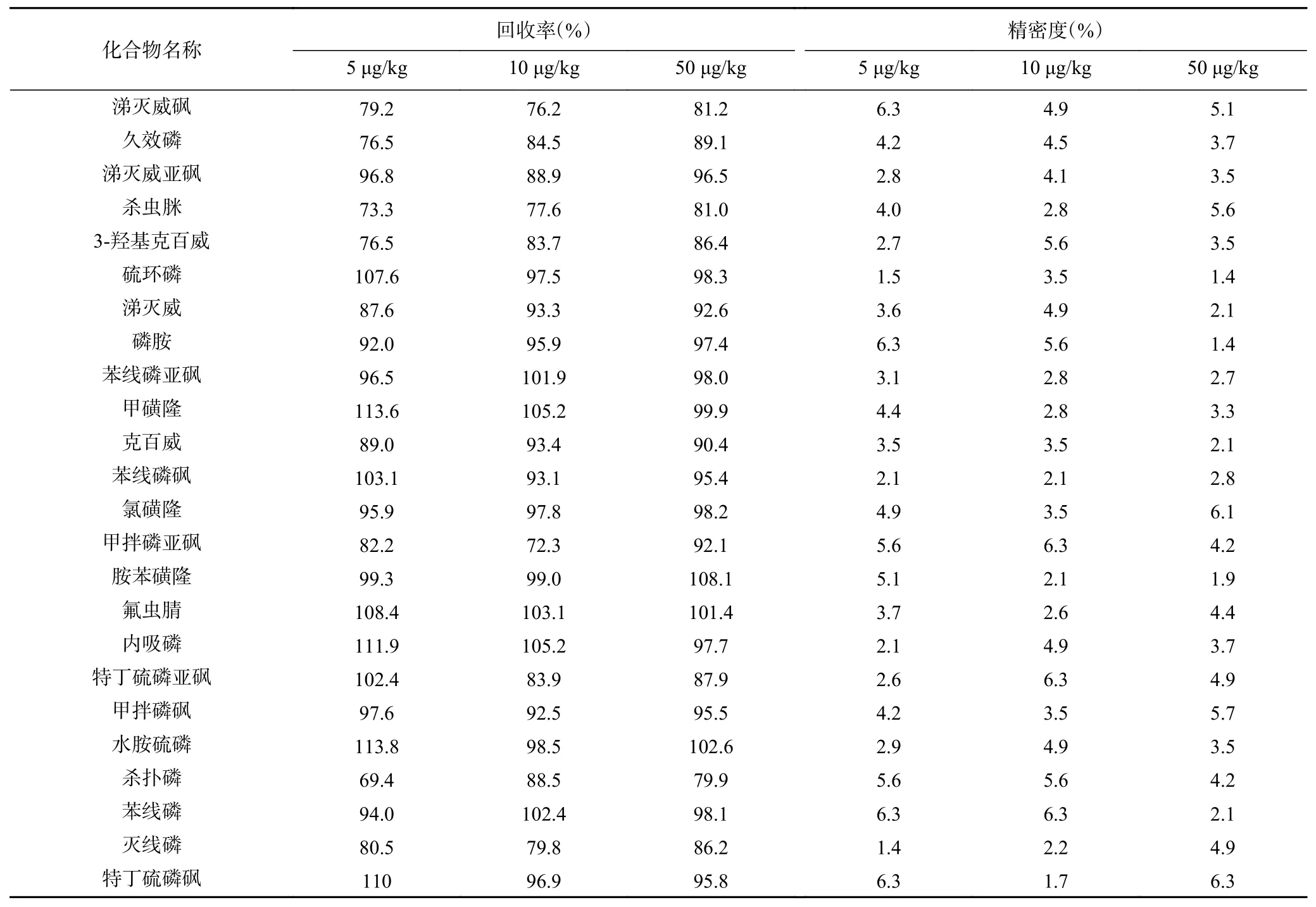
表3 加标回收和精密度的测定结果(n=6)Table 3 Determination results of recovery and precision (n=6)
2.7 实际样品的检测
采用本方法测定乳品企业样品9 份,配送鲜乳样品9 份,均未检出32 种农药残留。
3 结论
本研究结合固相支撑液液萃取在水系样本有机物萃取方面的优势,建立了生乳中32 种农药残留的超高效液相色谱-串联质谱快速检测方法。前处理过程无需活化,沉淀蛋白后上柱洗脱即可,15 min 内完成样品上柱与洗脱。相比于传统的液液萃取、固相萃取,简单快速,影响因素少,易于自动化。32 种目标物检出限为0.1~2.5 μg/kg,定量限为0.3~7.5 μg/kg,平均回收率为69.4%~113.8%,相对标准偏差小于8.2%,符合GB/T 27404 标准要求。以生乳基质为研究对象, 充实了动物源性食品中农药残留的检测方法,可为乳制品中农药残留的监管和风险评价提供技术支持。
