超高效液相色谱-串联质谱法测定猪尿中7种α2-受体激动剂残留
2023-08-23董文婷陈向丹李菁菁金秀娥
龚 波,王 峻,董文婷,陈向丹,李菁菁,金秀娥,周 平
(湖北省兽药监察所,武汉 430070)
以可乐定为代表的α2-受体激动剂在现代医学中常用于高血压的治疗,在兽医上广泛用于镇静和镇痛,也能加强麻醉药的作用和减少用量[1]。科学研究发现,该类药物可以通过作用于α2-受体,促进机体释放生长激素释放因子,调节磷酸腺苷水平,从而引起生长激素分泌增加[2-4]。从王雷杰[5]的动物实验中可以看出,饲喂添加可乐定的饲料能显著提高猪的瘦肉率,降低脂肪比率和背膘厚,改善猪的胴体组成。近年来,“瘦肉精”事件仍屡禁不止,时有发生,而且非法添加品种和手段不断升级,从盐酸克伦特罗、莱克多巴胺、沙丁胺醇等β-受体激动剂到可乐定等这类α2-受体激动剂。2010 年12 月27 日农业部1519 号公告已明确把可乐定列为《禁止在饲料和动物饮水中使用的物质》清单。因此,制定和完善α2-受体激动剂类药物多残留检测方法迫在眉睫。经查阅文献发现,对α2-受体激动剂类药物残留检测的研究主要集中于猪组织[6]、饲料[7]、血浆[8-9]、猪肉[10-11]、猪肝[12]、动物源食品[13-14]等基质中的可乐定等数种α2-受体激动剂含量检测。2004年,M.-H. Spyridak[15]报道了气相质谱法检测马尿中的赛拉嗪的方法;国家标准GB 31660.7-2019《猪组织和尿液中赛庚啶及可乐定残留量的测定 液相色谱-串联质谱法》以及孙晓亮[16]对猪尿中可乐定的残留检测进行了规定及研究。目前还未见以猪尿为基质,同时测定多种α2-受体激动剂药物的方法。本研究在不伤害猪本体的前提下取尿液样本,可快速、准确的同时测定7种α2-受体激动剂药物,填补检测方法空白,为实验室检测提供有效技术支撑。
1 材料和方法
1.1 仪器与耗材 超高效液相色谱-串联质谱仪Aglient 6470,配电喷雾离子源(ESI); MS3型涡旋混合器(德国IKA公司);pH计(上海雷磁公司,PHS-3E);冷冻高速离心机,转速≥10000 r/min(日立,CR21 N型);氮吹仪(美国Organomation公司,N-EVAP型);固相萃取装置(美国Agilent公司);MCX固相萃取柱(60 mg/3 mL,美国water公司);微孔滤头(0.22 μm有机相,美国Agilent公司)。
1.2 药品与试剂 赛拉嗪、盐酸可乐定、盐酸替扎尼定、溴莫尼定、胍法辛、胍那苄、盐酸安普乐定含量均≥90.0%,除胍法辛、胍那苄购于First Standard外,其余对照品均购于CATO公司;甲醇、乙腈(色谱纯,Tedi公司);甲酸(色谱纯,J.T.Baker,88%); 氢氧化钠、乙酸乙酯、氯化钠、氨水均为分析纯(国药集团化学试剂有限公司);实验室用水为超纯水。
1.3 实验方法
1.3.1 样品的前处理 取试样5.00 mL,于50 mL塑料离心管中, 用5 mol/L氢氧化钠溶液调pH值至10.0~11.0,涡旋混匀,加乙酸乙酯10 mL,加1.5~2.0 g氯化钠,充分振荡,于10000 r/min离心5 min,转移上清液于另一50 mL塑料离心管中。下层溶液中加乙酸乙酯10 mL重复提取一次,合并两次上清液,50 ℃氮气吹干,加0.1 %甲酸:甲醇溶液(90∶10)5.0 mL溶解残余物,备用。
取MCX固相萃取柱依次用甲醇3 mL、水3 mL活化,把全部备用液以每秒一滴的速度过柱,依次用水3 mL、甲醇3 mL分别淋洗,真空抽干,5 %氨水甲醇溶液5 mL洗脱,收集洗脱液,于50 ℃水浴中氮气吹干。用0.1%甲酸:甲醇溶液(90∶10)1.0 mL溶解残余物,涡旋1 min,过0.22 μm微孔滤头,供液相色谱-串联质谱测定。
1.3.2 液相色谱条件 色谱柱:C18柱,粒径2.7 μm,内径2.1 mm,柱长100 mm,或相当者;柱温35 ℃;流速0.3 mL/min;进样量10 μL;梯度洗脱程序延迟时间:1 min。梯度洗脱条件见表1。

表1 梯度洗脱程序Tab 1 Gradient elution program
1.3.3 质谱参考条件 离子源:电喷雾离子源(ESI);扫描方式:正离子扫描;毛细管电压(Capillary Voltage):4000 V;雾化温度(Gas temp):325 ℃;雾化器流速(Gas flow):10 L/min;雾化器压力(Nebulizer):40 psi;鞘气温度(Sheath gas temp):300 ℃;鞘气流速(Sheath gas flow):11 L/min;监测模式:多反应监测(MRM采集模式)7种α2-受体激动剂的MRM参数见表2。
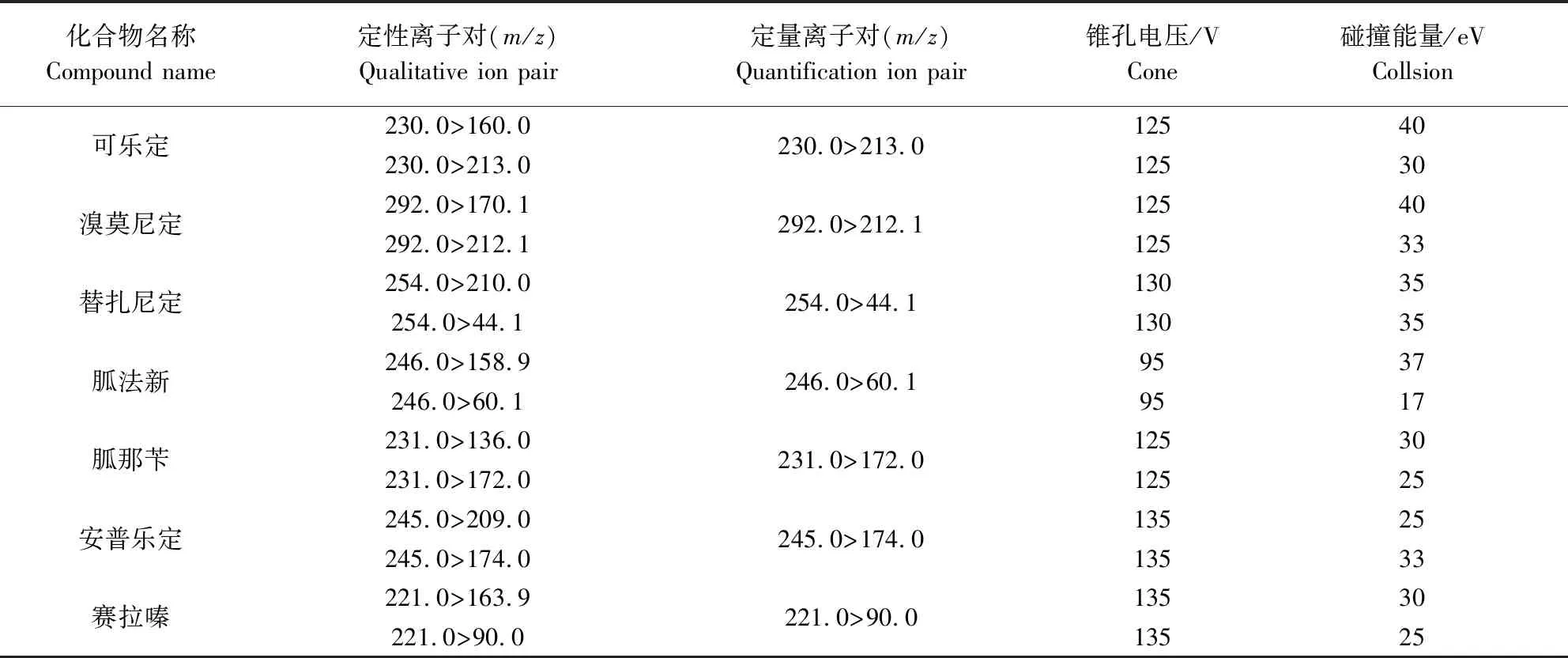
表2 7种α2-受体激动剂MRM参数Tab 2 MRM parameters of seven α2-agonists
1.3.4 标准溶液的配置 标准储备液:分别精密称取可乐定、溴莫尼定、替扎尼定、胍法辛、胍那苄、安普乐定、赛拉嗪7种对照品,用甲醇分别配制成1 mg /mL标准储备液,于2~8 ℃ 以下温度保存,有效期6个月。
混合标准中间液:分别准确量取1.00 mL可乐定、溴莫尼定、替扎尼定、胍法辛、胍那苄、安普乐定、赛拉嗪标准储备液(1.3.1项)置于100 mL量瓶中,用甲醇配制成浓度为10 μg/mL的混合标准中间液,于2~8 ℃以下温度保存,有效期1个月。
基质匹配标准曲线的构建:取空白样品按1.3.1项处理后作为基质空白溶液。准确移取适量混合标准中间液,用基质空白溶液配制成1、5、10、20、50、80 ng/mL 基质匹配标准工作液,供超高效液相色谱-串联质谱测定,现用现配。
1.4 基质效应测定 按照1.3.4项方法配制基质匹配标准曲线,同时准确移取适量的混合标准中间液,用0.1%甲酸:甲醇溶液(90∶10)配制成同样浓度的溶剂标准曲线,分别上机测试。基质效应计算公式为:基质效应(ME)=(基质匹配标准曲线斜率/溶剂配制标准曲线斜率-1)×100%。
2 结果与分析
2.1 标准曲线 用超高效液相色谱-串联质谱测定基质匹配系列标准工作液,7种α2-受体激动剂在1~80 μg/L浓度范围内均呈良好线性关系,线性方程及相关系数如表3所示。
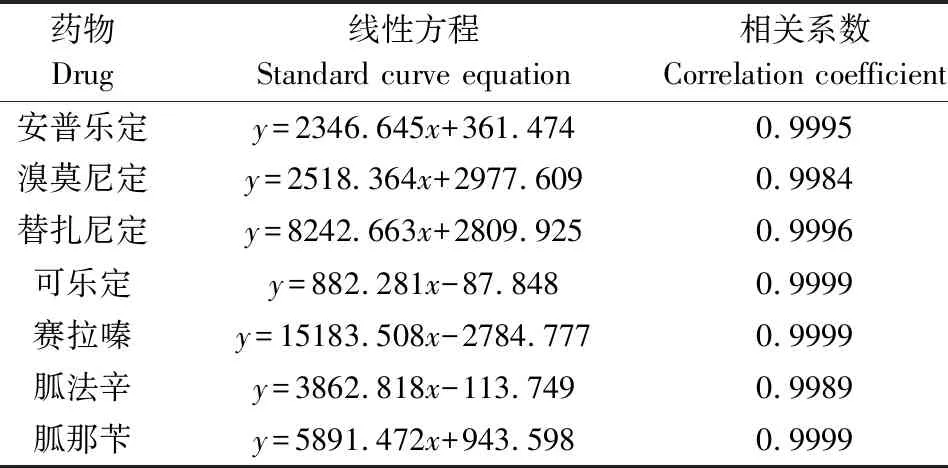
表3 基质匹配标准曲线方程Tab 3 Matrix matching standard curve equation
2.2 准确度与精密度 为考察方法的准确度和精密度,进行了添加回收试验。按照上述处理和测定方法,选取了猪尿液空白样品,各添加3个不同浓度(即0.3、0.6、3 μg/L),每个水平设3个平行,重复试验3批。求每个样品的回收率和批内,批间相对标准偏差。其中猪尿空白样品添加回收试验测定结果见表4。该实验表明,动物尿液中7种α2-受体激动剂平均回收率为60%~110%,精密度小于15%,说明该方法具有较好的准确度和精密度。图1、图2为空白猪尿及添加回收率质谱图。
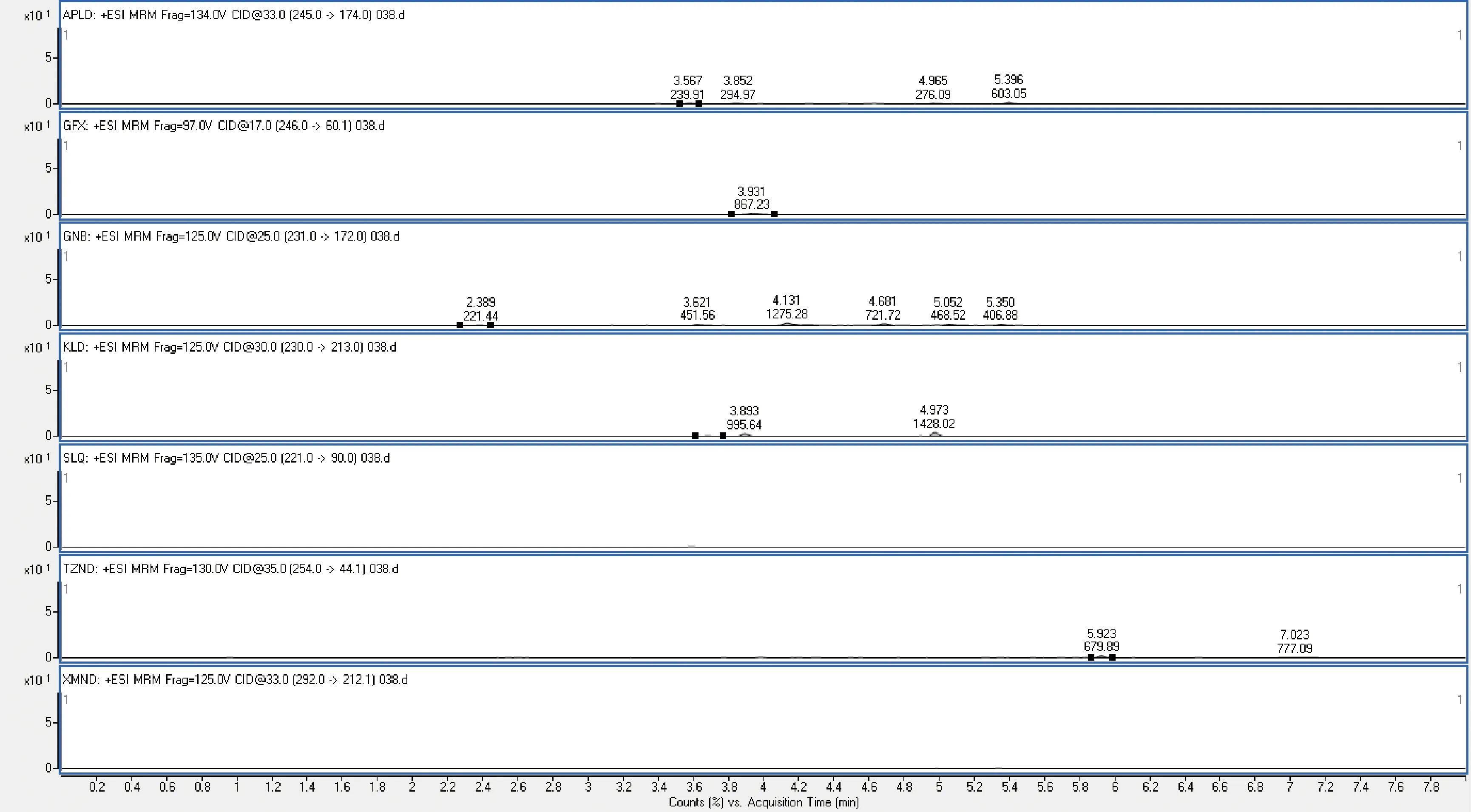
图1 空白猪尿样品的特征离子质谱图Fig 1.Characteristic ion mass chromatogram of blank pig urine

图2 猪尿中七种α2-受体激动剂添加回收试验的特征离子质谱图(3 μg/L)(图中依次为安普乐定、胍法辛、胍那苄、可乐定、赛拉嗪、替扎尼定、溴莫尼定)Fig 2.Characteristic ion mass chromatogram of seven α2-agonists added in blank pig urine(3 μg/L)(The order in the diagram is Apraclonidine、Guanfacine、Guanabenz、Clonidine、Xylazine、Tizanidine、Brimonidine)
2.3 检测限和定量限 猪尿液中7种α2-受体激动剂检测限为0.1 μg /L,信噪比S/N(Peak to Peak)>3;方法定量限为0.3 μg /L,信噪比S/N(Peak to Peak)>10。
2.4 基质效应的研究 对基质效应进行了研究,结果见表5。一般认为|ME|<20% 为弱基质效应, 20%≤|ME|≤50%为中等程度基质效应,|ME|>50%强基质效应。结果表明,部分待测物存在中等程度基质抑制效应,为降低基质效应对定量结果所的影响,采用基质匹配标准曲线外标法进行定量分析。
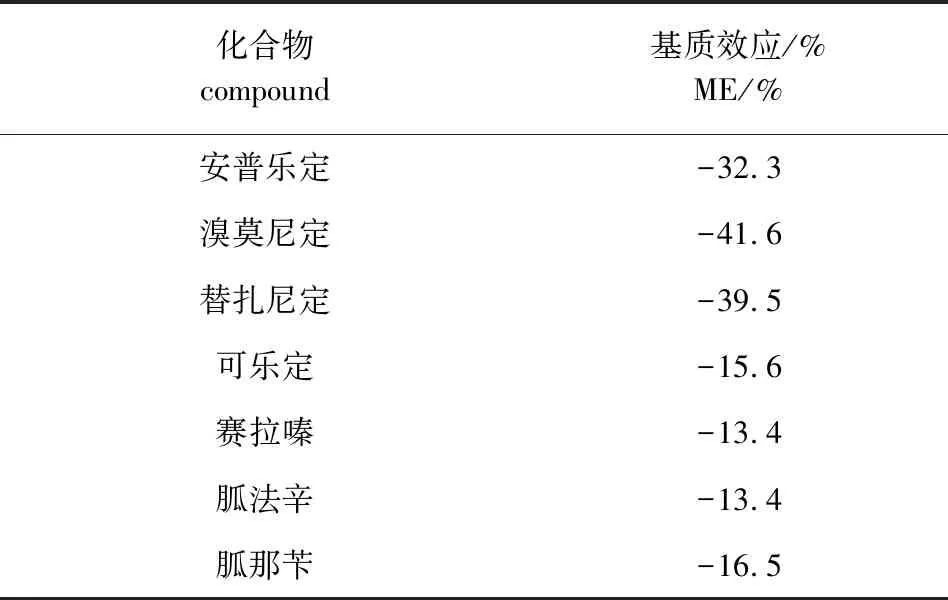
表5 7种α2-受体激动剂的基质效应Tab 5 ME of seven α2-agonists
2.5 缓冲液pH值的研究 本实验选择在碱性条件下进行样品提取,分别用5 M氢氧化钠溶液调节样品pH至8.0,9.0,10.0,11.0,11.5,12来进行比较,结果见表6。
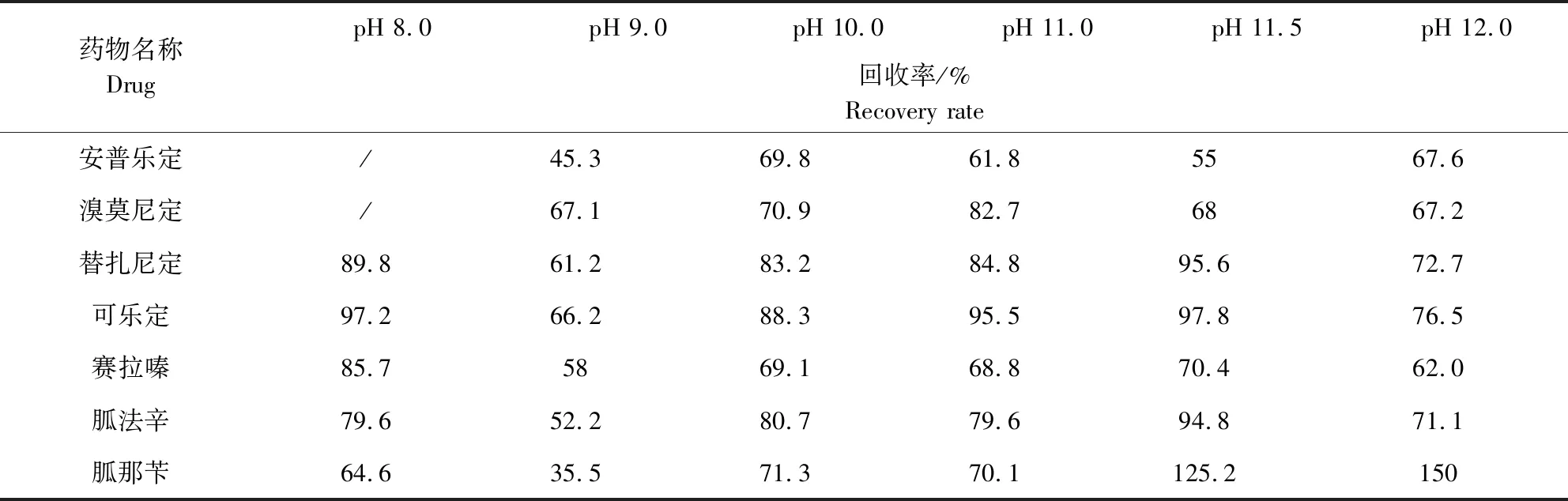
表6 不同pH值对添加回收率的影响Tab 6 The influence of different pH value on the recovery rate
由表6可以看出,当样品pH为8.0时,安普乐定、溴莫尼定峰形杂乱,无法定性、定量;当pH为9.0时,多种待测物添加回收效果不理想;当pH为11.5、12时,胍那苄出现不同程度的基质增强作用,导致添加回收率超过120%;而当pH范围为10.0、11.0时,样品峰形、回收率等都能达到理想效果。
3 讨论与结论
3.1 色谱条件的确定 色谱柱的选择:考察了常见的色谱柱ZORBAX SB-C18和Poroshell120 SB-C18对色谱峰进行分离,比较分离度、响应值以及峰形。通过比较发现,利用ZORBAX SB-C18可以获得较好的分离度,但是部分目标物会产生拖尾的色谱峰。Poroshell120 SB-C18均可以使7种待测物在5分钟内快速出峰,由于该色谱柱的填料孔径为2.7 μm,所以峰形也会比较窄,不会产生拖尾峰或者宽峰。考虑到方法的广泛适用性,最终推荐C18柱 (2.1×100 mm,2.7 μm),或相当者。
流动相的选择:α2-受体激动剂具有一定碱性特征,在酸性流动相体系中易产生稳定的[M+H]+离子,因此考虑常用的0.1%甲酸水作为流动相水相。为了避免溶剂效应,流动相有机相初始比例和样品复溶液有机相比例保持一致,选用0.1%甲酸水-甲醇的流动相体系,同时采用流动相梯度变化保证目标物有效分离。
3.2 质谱条件的优化 七种α2-受体激动剂均含有胺类结构,且极性较强,实验中选择正离子(ESI+)模式。配制100 ng/mL的标准溶液,采用不经过色谱柱直接连接质谱的方式,在全扫描中找出每个化合物的母离子,利用MRM采集模式优化子离子、碰撞电压、碰撞能量等参数。
3.3 样品前处理方法的优化
3.3.1 缓冲液pH值的优化 由于α2-受体激动剂具有一定碱的特性,在水环境中存在一定的电离现象,从而导致了有机试剂提取不完全。为了有效抑制其离子化的现象,本实验的缓冲液适合选择碱性环境。随机选取20份猪尿液样品,测得其pH范围为5.6~8.1,若直接加入一定量的碱性溶液[10-13],并不能保证样品提取溶液pH的稳定性,故本实验采用5 mol/L氢氧化钠溶液调pH值至10.0~11.0的方式保持提取的碱性环境。
3.3.2 提取试剂的选择 对样品中α2-受体激动剂的提取液进行了摸索,采用了常用的乙酸乙酯、乙腈提取溶剂。通过比较,乙腈提取效率不如乙酸乙酯提取,并且当用乙腈提取时,胍法辛基质干扰明显,故选用乙酸乙酯作为提取试剂。
3.3.3 样品净化过程优化 α2-受体激动剂属于碱性药物,故考虑对碱性药物具有高选择性的混合型阳离子小柱进行固相萃取净化。因样品经乙酸乙酯提取、氮气吹干复溶、高速离心后即可得到较为清亮的液体,我们选择了3种方法进行比较,一为样品经提取操作后不经过小柱净化直接离心取上清液上机,二为参考李艳明[11]李丹妮[6]分别采用PRIME HLB、QuEChERS净化后上机,三为样品经提取后用混合型阳离子小柱净化后上机。实验显示,方法二样品经净化后采集到的质谱图与方法三相当,但部分上机液体呈现淡黄色,不如方法三净化后透亮澄清,且PRIME HLB、QuEChERS成品价格较高,若采用填料自己填充又比较麻烦延长实验操作时间。而方法一采集到质谱图杂质干扰较大,长期操作会对液质联用仪造成污染及损害。从净化效果、经济实用等多方面考虑,此步骤选择用常用的混合型阳离子小柱净化。
3.3.4 样品复溶液的选择 考虑复溶液与流动相的一致性,分别比较了乙腈:0.1%甲酸水溶液=20∶80,甲醇∶0.1%甲酸水溶液=20∶80,乙腈∶0.1%甲酸水溶液=10∶90,甲醇∶0.1%甲酸水溶液=10∶90,甲醇∶0.1%甲酸水溶液=5∶95等5种溶剂作为最终进样前的样品复溶液。发现当复溶液为乙腈∶0.1%甲酸水溶液=20∶80时,安普乐定和溴莫尼定峰型较差,不符合定性定量要求,同时当有机相为乙腈时,液质响应普遍没有甲醇作为有机相时高。而当有机相为甲醇时,过高的甲醇浓度反而会抑制待测物的响应。当甲醇比例为5%、10%时,均能得到较为满意的液质图谱,因甲醇比例为10%时的待测物响应略高于5%甲醇,故最终确定样品复溶液为甲醇∶0.1%甲酸水溶液=10∶90。
本研究建立了一种适用于猪尿液中α2-受体激动剂类药物的超高效液相色谱串联质谱的检测技术。该方法在碱性环境中用乙酸乙酯提取,混合型阳离子柱净化,结合基质匹配标准曲线定量,无需采用内标校正[9],具有提取效率高,灵敏度高,检测成本低的特点,满足了检测要求,弥补了暂无猪尿中多种α2-受体激动剂类药物检测方法的行业空白。
