单增李斯特菌感染RAW264.7 细胞转录组差异表达分析
2023-08-22马红梅马臣杰马玲玲马嘉琦
马红梅, 马臣杰, 岳 苑,3, 马玲玲, 马嘉琦, 曾 瑾*
(1. 宁夏大学 西部特色生物资源保护与利用教育部重点实验室,宁夏 银川 750021;2. 宁夏大学 生命科学学院,宁夏 银川 750021;3. 宁夏回族自治区食品检测研究院,宁夏 银川 750002)
单核细胞增生李斯特菌(Listeria monocytogenes,LM),简称单增李斯特菌,是一种常见的革兰氏阳性胞内寄生菌。 该菌为短杆菌,大小为0.5 μm×(1.0~2.0) μm,兼性厌氧、嗜冷、不含芽孢,不形成荚膜,广泛存在于周围环境中[1-3]。 LM 是一种人畜共患致病菌,主要由污染的食物传播,是世界卫生组织列举的四大食源性病原菌之一[4-7]。 据国家卫健委报道,LM 导致的食源性疾病病例数在全国食源性病例数中占很大比例。 LM 是李斯特菌众多类型中唯一能引起人类李斯特菌病(Listeriosis)的主要病原体[8],尤其容易感染孕妇、新生儿等免疫力低下的人群[9-11]。即使是低水平的食物污染也有可能导致局灶性感染、 细菌性败血症和脑膜炎以及胎儿感染,导致流产或妊娠并发症[12-13],甚至引起危及生命的疾病[14-15]。 据美国疾病预防控制中心报道,美国每年患有李斯特菌病的人数多达2 000 人,死亡率高达30%,其致死率甚至高过其他流行性致病菌,如沙门氏菌及肉毒杆菌[16]。据报道,恶性肿瘤或器官移植患者更易患侵袭性李斯特菌病, 人类免疫缺陷病毒(HIV)阳性患者侵袭性李斯特菌病的发病率是普通人群的500~1 000 倍[17]。目前,由于其高病死率[18-19]以及高耐药性,李斯特菌病仍是许多国家关注的重大公共卫生问题和面临的主要挑战[20-24]。
目前关于LM 的研究主要在LM 的检测以及应用上, 但关于LM 对机体的致病机制研究尚不明确[25-26]。 因此作者对LM(ATCC 19115)感染前后 的受体细胞RAW264.7 进行了转录组测序分析[27],期望从LM 感染前后的受体细胞中找到差异表达的基因和代表性的差异表达通路,从而为进一步研究LM 的致病机理提供理论基础。
1 材料与方法
1.1 主要材料
小鼠单核巨噬白血病细胞(RAW264.7)和单核细胞增生李斯特菌(ATCC 19115):均为作者所在实验室保存。
1.2 单核细胞增生李斯特菌的培养
将保有LM 的磁珠在脑心浸液 (brain heart infusion,BHI)固体培养基上划线接种,37 ℃恒温培养16~18 h 后挑单菌接至10 mL BHI 液体培养基中,37 ℃、180 r/min 振荡培养至OD600达0.6~0.8。吸取细菌培养液至无菌的1.5 mL 离心管,12 000 r/min 离心30 s 收集细菌,无菌PBS 洗涤3 次,用1 mL DMEM 完全培养基重悬混匀后, 测定OD600,根据需要调整菌液的浓度。
1.3 RAW264.7 细胞培养及感染
RAW264.7 细胞培养在由基础培养基和体积分数10%新生小牛血清(newborn calf serum,NBCS)组成的DMEM 完全培养基中, 取对数期RAW264.7细胞铺于6 孔细胞培养板, 每孔细胞数量为1×106个, 在37 ℃和体积分数5%的CO2条件下过夜培养,细胞密度达到约80%时进行处理。 本实验主要分为2 组,每组进行3 次重复,LM 感染组(MOI=10)命名为NL1、NL2 和NL3,感染时间为6 h;另外一组未处理的细胞作为对照组, 命名为NC1、NC2 和NC3。
1.4 总RNA 提取
使用Trizol 试剂(TIANGEN)提取细胞总RNA,Nanodrop8000 测定RNA 浓度。 OD260/OD280在1.8~2.2 为合格样本。
1.5 文库构建及测序
RNA-seq 实验过程涉及样本检测、 富集mRNA、 合成cDNA 第一条链和第二条链、 末端修复、 加A 尾和接头、 筛选cDNA、 PCR 扩增、 纯化PCR 产物、建库。文库质检合格后测序,流程见图1。
1.6 数据质控
测序原始数据经过一系列过滤、筛选以及错误率检查、GC 含量分布检查,最后获得clean reads[28]。 后续所有分析均为对clean reads 数据进行分析所得[29]。
1.7 差异基因筛选
使用DESeq2(Anders et al,2014)软件进行差异表达分析,分析方法基于负二项分布。 以P<0.05 以及|lb F|≥1.5(其中F 为fold change)为标准筛选差异表达基因(differentially expressed genes,DEGs)[30]。
1.8 差异基因富集分析
对筛选后符合标准的DEGs 进行GO(Gene Ontology,GO) 和 KEGG (Kyoto Encyclopedia of Genes and Genomes,KEGG)富集分析[31]。利用GO 对注释到的基因功能进行分析,利用KEGG 根据参与的通路或功能对DEGs 进行分类和统计。 富集分析标准为P<0.05。
1.9 RT-qPCR 验证
作者选取了11 个DEGs 对分析结果进行RTqPCR 验证。 对各基因扩增结果计算相对表达量,并与RNA-Seq 结果进行比较。
1.10 统计学分析
对计算结果进行统计学分析, 绘图软件为GraphPad Prism 8, 数据结果表示为Mean±SD (P<0.05 表示差异显著,P<0.01 表示差异极显著)。
2 结果与分析
2.1 数据质控分析
对LM 感染组和对照组获得的clean reads 进行汇总,见表1。Q20 大于95%,Q30 大于90%,表明经LM 感染后的RAW264.7 细胞所提取的RNA 质量良好,转录组测序结果可信。
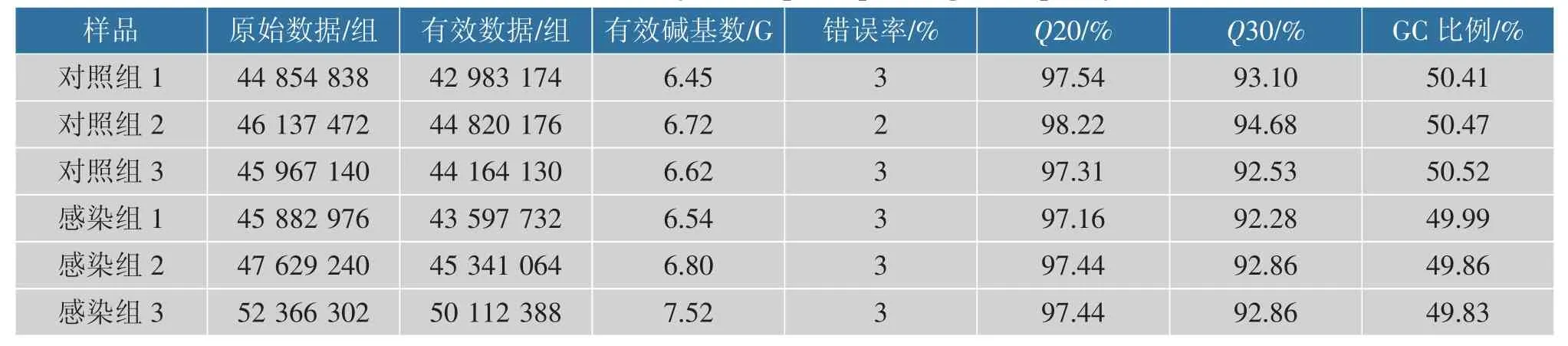
表1 样本测序数据质量汇总Table 1 Summary of sample sequencing data quality
2.2 样本间相关性分析
相关性分析可用于检验实验可靠性和样本合理性, 相关系数值越接近1 代表样本的相似度越高。 生物学重复样品间R2一般要求大于0.8。 由图2 可知,本研究中的3 次独立重复实验所收获的细胞RNA 样品经转录组测序后, 样本间相关性系数均大于0.8, 证明3 次重复实验的样本间差异不显著,因此后续的转录组测序结果差异分析可靠。
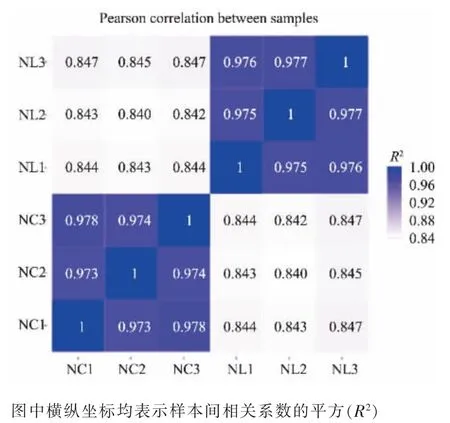
图2 样本间相关性分析热图Fig. 2 Heat map of correlation between samples
2.3 差异基因筛选
edgeR 软件筛选出P<0.05 以 及| lb F |≥1.5 的DEGs(其中F 为基因表达倍数变化)。图3 显示,LM感染RAW264.7 细胞后,DEGs 为1 782 个,其中上调的DEGs 为989,下调的DEGs 为793,筛选结果见火山图。 另外将DEGs 取并集后进行分析[28],表达相似基因均可聚类在一起,见图4。
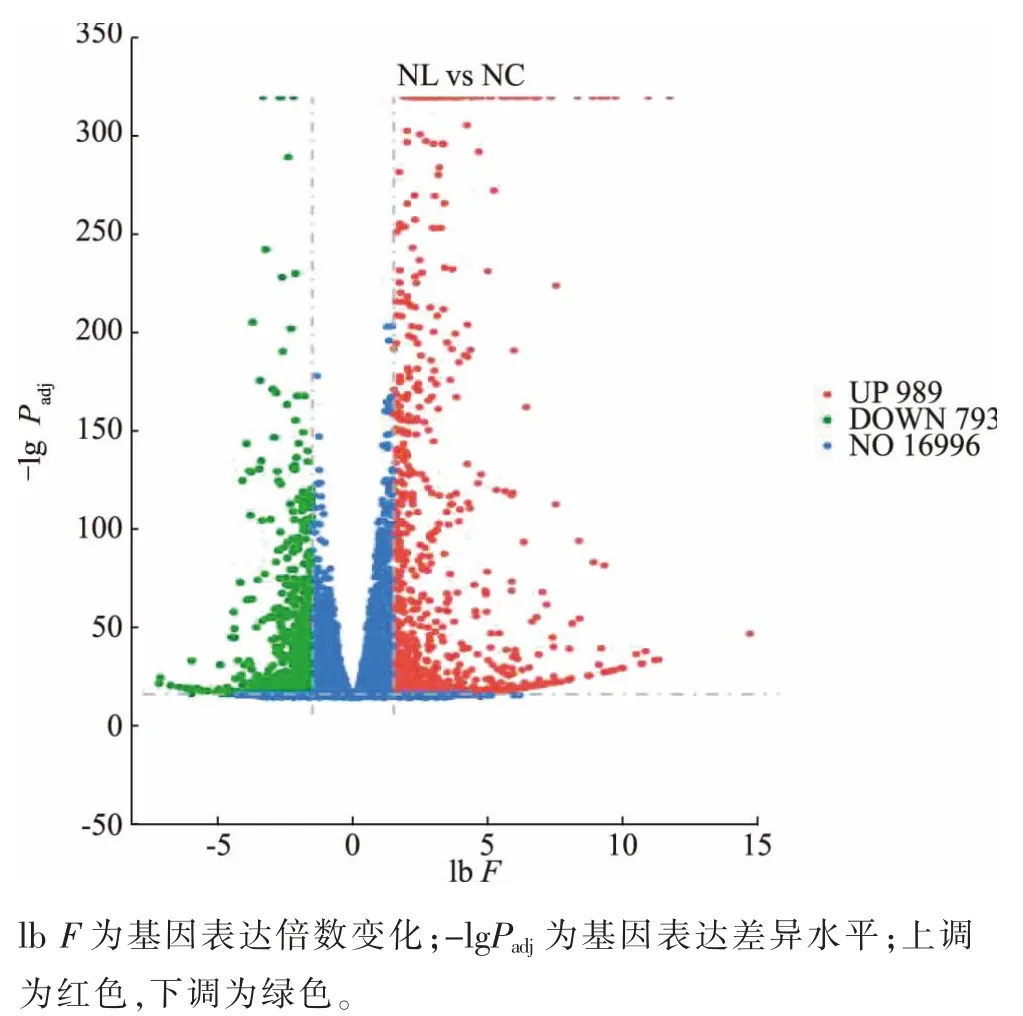
图3 差异基因火山图Fig. 3 Differential gene volcano map

图4 差异表达基因聚类热图Fig.4 Clustering heat map of differentially expressed genes
2.4 差异基因GO 功能富集分析
对聚类后的DEGs 进行GO 功能富集分析,共得到617 条GO 注释,其中51.05%(315 条)涉及生物学过程 (BP),36.79%(227 条) 涉及分子功能(MF),12.16%(75 条) 涉及细胞组分(CC)。 图5 为前30 条注释结果,BP 中DEGs 显著集中在免疫系统过程(immune system process)、免疫反应(immune response)、G 蛋白偶联受体信号通 路 (G-protein coupled receptor signaling pathway)、 细胞内信号转导(intracellular signal transduction)、细胞程序性死亡的调控(regulation of programmed cell death)等;CC 中DEGs 显著集中在胞外区(extracellular region) 中;MF 中DEGs 显著集中在细胞因子活性(cytokine activity)、 细 胞 因 子 受 体 结 合(cytokine receptor binding)、信号受体结合(signaling receptor binding)、 分 子 功 能 调 节 (molecular function regulator)、受体调节活性(receptor regulator activity)和受体配体活性(receptor ligand activity)等。在所有注释中,细胞因子活性最显著。
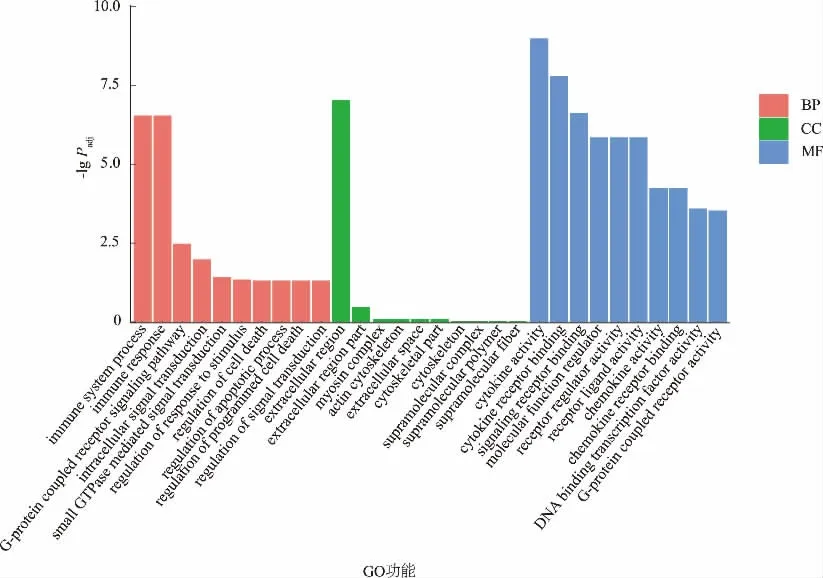
图5 GO 富集分析柱状图Fig. 5 Histogram of GO enrichment analysis
2.5 差异基因KEGG 富集分析
对DEGs 进行KEGG 分析,DEGs 富集到了304条信号通路中,显著性变化的有44 条(P<0.05)。 图6 为富集的前19 条KEGG 通路[28],DEGs 在肿瘤坏死因子信号通路(TNF signaling pathway)、甲型流感(Influenza A)、IL-17 信 号 通 路 (IL-17 signaling pathway)、NF-κB 信号通路 (NF-kappa B signaling pathway)等信号通路显著富集。 对显著富集的前几条信号通路的差异基因进行统计, 结果见表2。KEGG 通路分析表明, 上调的基因主要集中在促炎细胞因子、趋化因子等免疫调控方面的基因,例如白介素IL-6、IL-1β、TNF、NLRP3、CC 基序趋化因子CCL2 和CCL22 等。
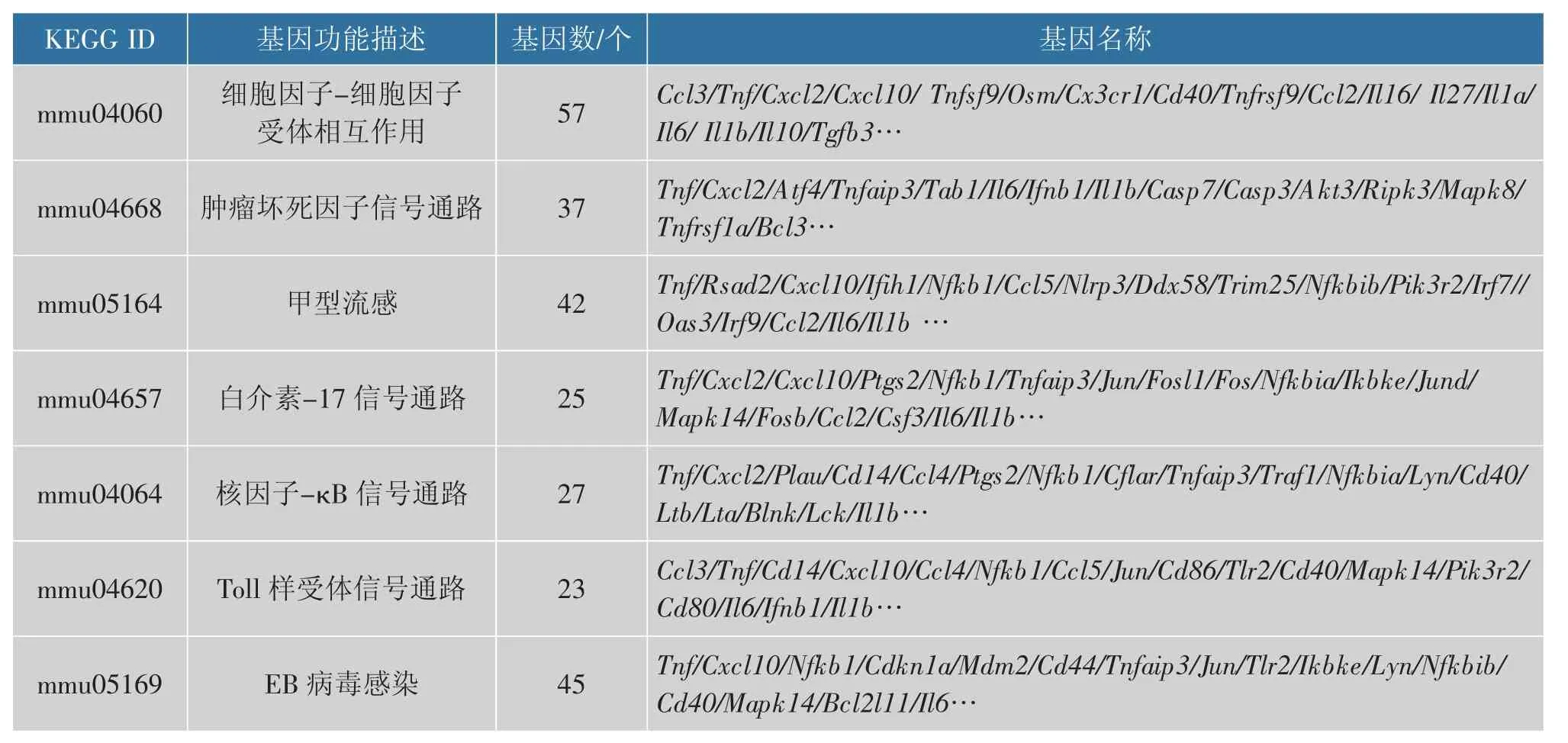
表2 显著富集信号通路及其差异表达基因Table 2 Significantly enriched signaling pathways and their differentially expressed genes
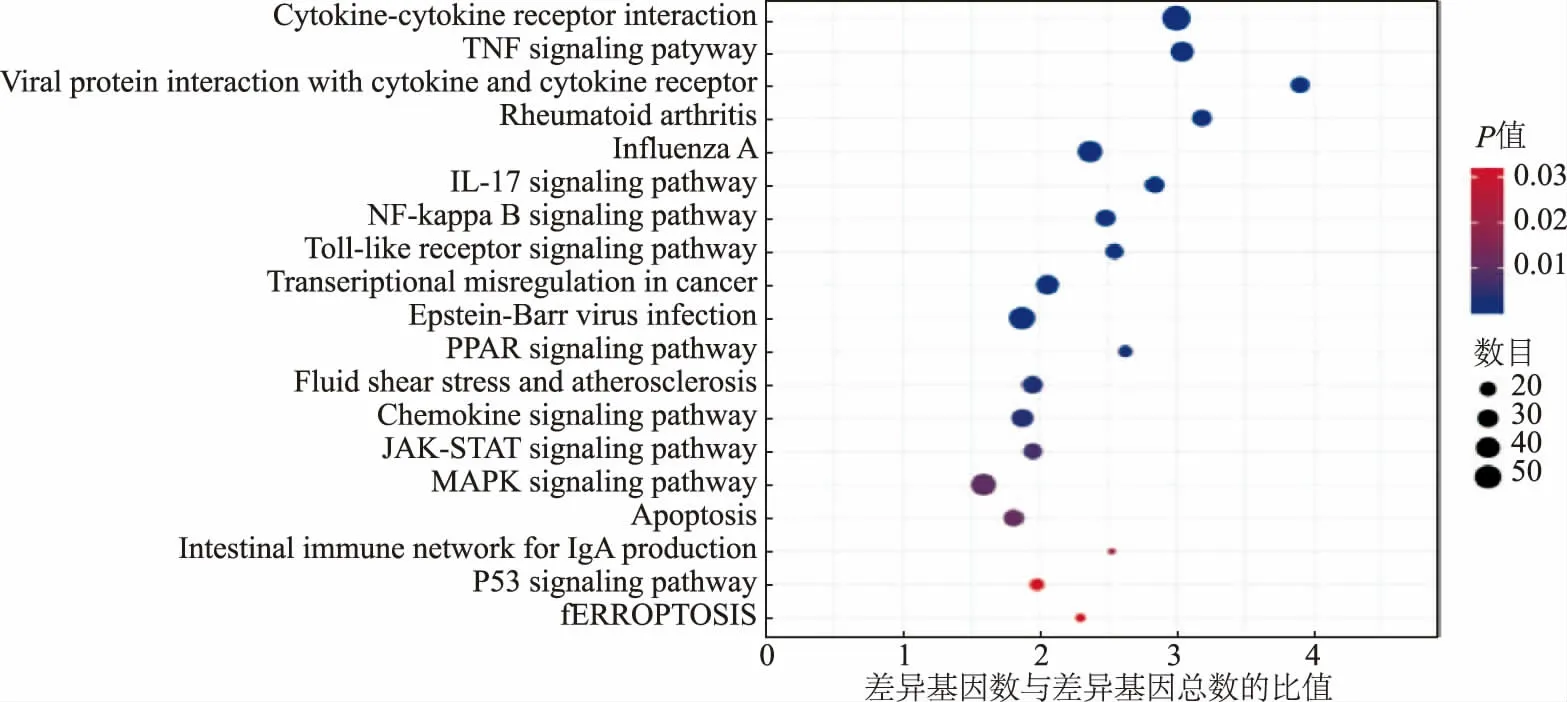
图6 前19 条富集的KEGG 信号通路散点图Fig. 6 Scatter diagram of the first 19 enriched KEGG signaling pathways
2.6 RT-qPCR 验证
为了进一步验证结果准确性,从表2 列出的通路中筛选出11 个基因(5 个上调,6 个下调)进行验证,RT-qPCR 引物序列见表3,对比结果见图7。 由图7 可知,RT-qPCR 结果与转录组测序一致, 表明数据可靠。
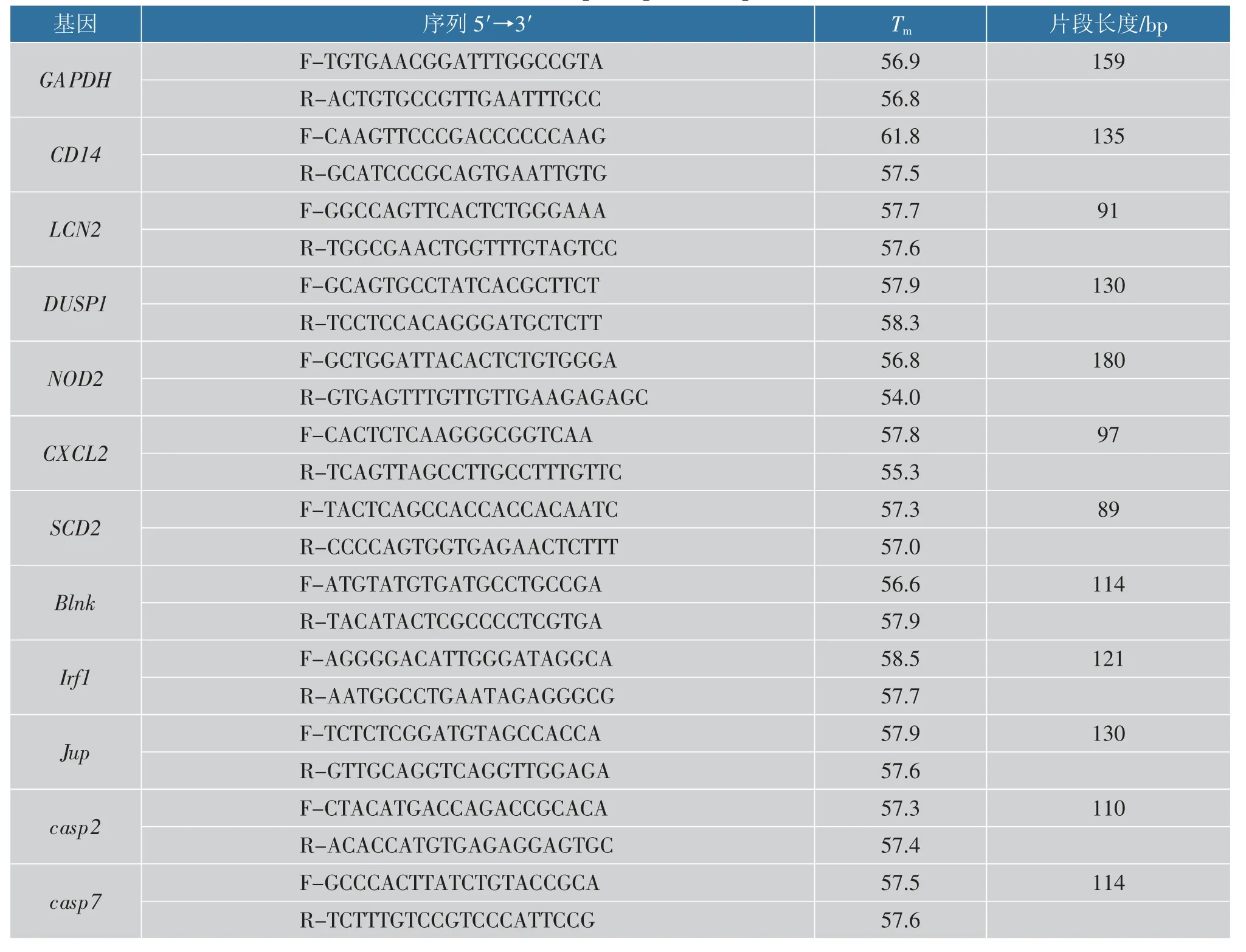
表3 RT-qPCR 引物序列Table 3 RT-qPCR primer sequence
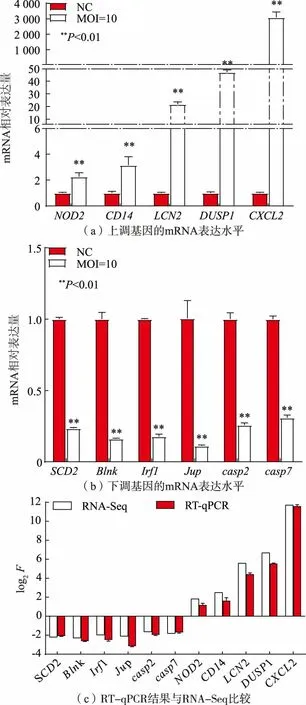
图7 RT-qPCR 数据与RNA-Seq 数据比较Fig. 7 Comparison of RT-qPCR data and RNA-Seq data
3 讨 论
李斯特菌病是许多国家关注的重大公共卫生安全问题,在2017 年6 月中旬,南非曾出现李斯特菌病病例爆发的案例。 据美国国家传染病研究所(NICD)报告,在1 060 个实验室检测确认的李斯特菌病病例中死亡人数多达216 人,通过流行病学调查发现, 爆发的主要原因是食用了被LM 污染的即食肉制品。 通过对患者的不同分离株进行MLST(multilocus sequence typing,MLST)分析[32],发 现 来自患者的分离株中很大一部分(91%)的序列类型为6 型 (ST6)[33-35]。 Zhang 等使用脉冲场凝胶电泳(PFGE)和全基因组测序(WGS)对上海市即食肉类加工厂中的单核细胞增生李斯特菌进行了研究,结果显示在48 个送检样本中,有12 个样本存在单核细胞增生李斯特菌分离株,其中ST5(1/2b)分离株占主导地位(83.3%,10/12)[20]。 另外,有研究团队对2008—2019 年来自29 个医疗机构的144 例李斯特菌病病例进行了分析,其中包括96 例母婴感染,33例菌血症,13 例神经性李斯特菌病和2 例皮肤性李斯特菌病。这些病例中,52 例感染孕妇有31 例胎儿流产 (占59.6%),41 例新生儿患者中有4 例死亡(占9.8%),通过MLST 分析将144 株临床分离菌株区分为23 个单独的ST 序列类型。 分析结果显示,最流行的ST 是ST87(49 株,占34.0%)。 此外,该研究发现所有单核细胞增生李斯特氏菌分离株对青霉素、氨苄青霉素和美罗培南均敏感,这也说明,导致临床李斯特菌病的单核细胞增生李斯特菌菌株具有基因多态性, 同时应该警惕ST87 和ST1 型所引起的高患病率[36]。
单增李斯特菌是一种胞内寄生菌,是动物和人类胃肠道的暂时性寄居者[37-38]。 它能穿越包括胎盘屏障、血脑屏障、肠上皮紧密连接在内的多种生理性屏障,能广泛定植于各种组织器官。 LM 细胞膜表面存在2 种重要的毒力因子InlA 和InIB,在LM 中通过肠上皮屏障和胎盘屏障发挥关键作用[39]。 溶血素O(listeriolysinO,LLO)是LM 分泌的成孔毒素,在LM 从囊泡逃逸进入胞浆内的过程中发挥重要作用[40-42]。 ActA 也是LM 的胞膜表面蛋白质,介导LM 在细胞内的迁移以及在相邻细胞和组织间的传播[43-44]。
在胃肠道内,先天免疫受体起着抵抗侵入性病原体的直接防御机制作用,而细菌建立感染的第一步,也是最重要的一步,就是微生物病原体可能通过毒素、RNA、DNA 作用于宿主细胞,触发宿主细胞内的信号转导,引起细胞生理和生化变化,同时宿主细胞也通过分泌某些蛋白质进行免疫调控[45-46]。这种细菌与宿主的相互作用不仅决定了疾病的转归,而且对理解病原微生物的早期感染机制也具有重要意义[47-49]。
转录组分析在真菌及细菌的致病机理研究中应用广泛。 唐光甫等人利用转录组分析,对红曲中桔霉素导致毒性的内在机理进行了探究[50],发现桔霉素可能通过TP53、CASP3、MAPK3 和ALB 等关键靶点导致肝肾毒性,揭示了桔霉素导致肝肾毒性的潜在作用靶点和信号通路,为进一步研究桔霉素致毒机理和健康防护提供理论参考。 目前还未有LM感染RAW264.7 细胞后转录组表达谱差异分析的报道。 RAW264.7 细胞是研究细菌感染的经典细胞系[51]。为了探讨RAW264.7 细胞经LM 处理后基因表达谱的变化, 用LM 感染RAW264.7 细胞6 h 后提取感染组和对照组细胞总RNA 进行转录组测序。 结果表明,与对照组相比,差异表达基因有1 782 个,其中上调为989, 下调为793。 对DEGs 进行GO 和KEGG 富集分析, 测序分析的结果表明DEGs 大多富集在与炎症相关的信号通路中,如肿瘤坏死因子信号通路、NF-κB 信号通路、 凋亡通路和IL-17 信号通路等。 对上述通路中高表达的基因进行研究,基因NLRP3 在LM 感染后显著上调。 NLRP3 也称NOD 样受体蛋白质3 (Nod-like receptor protein 3,NLRP3),它能结合凋亡相关斑点样蛋白(ASC)和天冬氨酸特异性半胱氨酸蛋白酶-1(caspase-1)形成NLRP3 (NLR family pyrin domain containing 3,NLRP3)炎性小体。 研究表明,NLRP3 在心血管疾病(CVD)[52]、动脉粥样硬化[53-55]、败血症[56]、糖尿病肾病[57-58]、帕金森病[59]、缺血性肌肉损伤[60]等多种疾病中发挥重要作用。本课题组也对NLRP3 进行了相关研究[61],NLRP3 炎性小体可由双信号模式激活,微生物或内源性细胞因子刺激细胞, 导致核因子κB(nuclear factor-kappa B,NF-κB)的激活,NF-κB 激活后则会介导IL-1β 和IL-18 等重要的细胞炎性因子前体的产生,同时NF-κB 激活NLRP3 mRNA 的转录,再进行翻译及翻译后修饰等, 此过程即称为NLRP3 priming, 也被认为是经典的NLRP3 炎性小体活化中的第一信号[62-63]。 第二信号可由ATP、毒素、病毒、ROS 等识别位于细胞质内的模式识别受体NLRs 激活[64-65]。 2 种信号也会共同作用,导致一系列严重的炎症反应[66-67]。 总之,结合转录组分析及以上研究数据表明, 在LM 感染RAW264.7 细胞中,NLRP3 介导的炎症反应可能发挥了极其关键的作用。
4 结 语
通过转录组分析发现,RAW264.7 细胞在感染LM 6 h 后差异表达基因有1 782 个, 其中上调为989,下调为793。 通过对基因注释发现,DEGs 主要集中在免疫系统过程、免疫反应、细胞内信号转导、细胞程序性死亡的调控、细胞因子活性、细胞因子受体结合、信号受体结合、分子功能调节、受体调节活性和受体配体活性等生物学功能。 除此之外,DEGs 大多富集在与炎症相关的信号通路中, 如肿瘤坏死因子信号通路、NF-κB 信号通路、Toll 样受体信号通路、凋亡通路和IL-17 信号通路等。 通过对功能基因进行分析,发现NLRP3 基因在LM 感染后显著上调,这为单增李斯特菌感染RAW264.7 细胞后介导的炎症反应提供了新思路, 也为进一步研究LM 的致病机理提供思路和研究基础。
