整合影像组学和基因组学构建肾透明细胞癌肿瘤分级预测模型
2023-08-21田占雨李霞李永生陈家齐
田占雨,李霞,李永生,陈家齐
海南医学院生物医学信息与工程学院,海南 海口 571199
肾透明细胞癌(clear cell renal carcinoma,CCRCC)占肾细胞癌的75%~80%,是恶性程度高,预后差,死亡率最高的亚型[1]。世界卫生组织/国际泌尿病理学会(World Health Organization/International Society of Urological Pathology,WHO/ISUP)根据核仁突出情况对CCRCC 进行了分级(I级、Ⅱ级、Ⅲ级、Ⅳ级),随后根据预后结局进一步将患者分为低级别组(I级、Ⅱ级)和高级别组(Ⅲ级、Ⅳ级)[2-3]。现阶段对肿瘤分级主要依靠经皮穿刺活检,需要面临出血、气胸以及针道转移等并发症的风险。因此,需要一种非侵入性的方法协助判断CCRCC的肿瘤分级。随着医学影像成像技术的发展以及高通量基因组学技术的进步,影像基因组学应运而生。影像基因组学是一个多学科交叉的新兴领域,是影像组学和基因组学的有机结合。在之前的研究中,Celli等[4]结合了影像组学与基因组学,利用多元逻辑回归算法构建机器学习模型,该模型在鉴别诊断低风险子宫内膜癌方面有着出色的分类能力。马佳琪等[5]通过结合影像基因组学信息,在诊断结直肠癌肝转移、预测患者预后和治疗敏感性等方面显示出独特的优势。将预测模型用于临床实践为个性化治疗带来显著优势,可以减少患者后续治疗的时间成本,并为制定临床治疗方案和改善患者总体生存率提供了指导和新的见解。然而,针对恶性程度较高的CCRCC的肿瘤分级仍然具有挑战性。
本研究以影像基因组学为切入点,通过结合医学影像和多组学数据识别不同水平的特征,基于机器学习算法构建多种预测模型,并结合各类算法优势,筛选最优特征组合以达到准确预测CCRCC肿瘤分级的目的。相较于之前的研究,本研究构建的复合型模型取得了更优的分类效能。此外,进一步探讨了多组学水平特征基因的功能,挖掘基因在CCRCC 中的潜在临床价值。
1 资料与方法
1.1 一般资料 CCRCC患者的医学影像数据来源于TCIA 数据库(The Cancer Imaging Archive,version 3:Updated 2020/05/29),按照入组标准进行筛选。入组标准:(1)经病理诊断确诊为CCRCC;(2)CT 增强扫描皮质期与髓质期期相准确且完整;(3)图像中未出现各类伪影;(4)具备合适的信噪比。符合入组标准的样本共197 例。CCRCC 患者的多组学数据来源于TCGA数据库(The Cancer Genome Atlas Program-NCI,2017年)。数据集包含了530例患者的基因组和转录组信息,基因表达水平为每千个碱基的转录每百万映射读取的片段数(fragment per kilobase method,FPKM)。通过比对注释文件,获得了16 769个编码基因的表达谱数据和12 903个基因的遗传变异数据。最后从数据集中筛选出同时具有医学影像数据和多组学数据的样本,包含了197例患者。随后本研究按照2016年WHO/ISUP简化分级将I期、II期定义为“低级别组(stage 0)”,III期、IV期定义为“高级别组(stage 1)”。
1.2 影像组学数据提取及预测模型构建 基于医学影像数据利用ITK-SNAP软件半自动形式确定原发灶边界,其中窗宽窗位预设为腹部窗(level:40 HU;window:600 HU)。随后应用Python软件(version 3.8)中“Pyradiomics”库提取影像组学特征[6],参数设定如下:箱宽(bin-width)为25;重采样像素间距(Pixel Spacing)为3 mm×3 mm×3 mm;内插程序选择最近邻。提取出的影像特征主要包含7 个类别维度,形状特征(shape-based)、一阶特征(first-order)、灰度共生矩阵(Gray-Level Co-occurrence Matrix,GLCM)、灰度区域大小矩阵(Gray Level Size Zone Matrix,GLSZM)、灰度行程矩阵(Gray Level Run-Length Matrix,GLRLM)、邻域灰度差矩阵特征(Neighbouring Gray Tone Difference Matrix,NGTDM)和灰度依赖矩阵(Gray Level Dependence Matrix,GLDM)。
1.3 识别与CCRCC 相关的转录组和基因组特征 在转录组数据集中,由于FPKM 值包含不同数量级,为避免表达值相差过大而对模型产生不良影响,本研究进行了数据的归一化,使基因表达谱数据局限在相同数量级范围内。由于转录组水平和基因组水平的数据形式不同,本研究分别进行了差异分析,以筛选在不同CCRCC肿瘤分级之间显著差异表达或突变的特征基因。
1.4 基因功能富集分析 为发掘在CCRCC肿瘤分级中显著差异表达的特征基因的潜在信息,探索基因的生物学功能以及肿瘤的生物学行为,本研究使用R 编程软件中“clusterProfiler”包中的“enrichGO”功能进行功能富集分析。对于富集分析得到的生物学功能,进一步筛选出至少包含3 个重合基因的显著富集的功能(count>3,P-value<0.05)。随后使用R“simplify-Enrichment”包中的“GO_similarity”功能进行语义相似性计算,将相似的GO术语进行聚类。最后使用“simplifyGO”功能对功能富集聚类结果进行可视化。
1.5 构建CCRCC肿瘤分级预测模型 本研究基于单一组学特征和联合多组学特征,并结合不同的机器学习算法构建了针对CCRCC 肿瘤分级的预测模型。主要基于支持向量机算法(Support Vector Machine,SVM)、逻辑回归算法(Logistic Regression,LR)、随机森林算法(Random Forest,RF)以及极端梯度提升算法(Extreme Gradient Boosting,XGB)分别构建机器学习模型,用于预测CCRCC 肿瘤分级。各个模型模型参数均采用网格搜索选取最佳输入参数,并且超参数均位于对应值阈内。
1.6 统计学方法 本研究通过Wilcoxon 秩和检验筛选在CCRCC高低分级中存在显著差异的影像组学特征和显著差异表达基因(P<0.05)。在基因组层面,本研究应用Fisher 精确检验筛选存在显著差异的突变基因(P<0.05)。最后,针对全部种类的特征应用最小绝对收缩和选择算子(least absolute shrinkage and selection operator,LASSO),筛选出具备较高权重的关键特征。
2 结果
2.1 CCRCC 影像组学的特征 本研究利用Python 软件中Pyradiomics 库,提取了197 例入组样本的107个影像组学特征。随后经过Wilcoxon秩和检验筛选[7],得到在CCRCC 不同分级之间存在显著差异的24 个影像组学特征(图1)。这些特征共同描述出肿瘤的形态和特点(图2),如肿瘤表面积体积比(original shape surface volume ratio),比值越低则说明肿瘤越近似于紧凑的球状;平均值(original first order mean)是评估ROI 内的平均灰度,集群阴影(original GLCM cluster shade)是衡量灰度共生矩阵(Gray-Level Co-occurrence Matrix,GLCM)的偏度和均匀度的指标,集群阴影的特征值越高则图像中平均值的不对称性就更高,这表示原发灶的内肿瘤组织可能出现出血或坏死。按照CCRCC不同的肿瘤分级绘制了各影像组学特征的表达值。网格体积(original shape mesh volume)应用三角形网格计算肿瘤原发灶体积,在高级别组中网格体积特征值较高,这表明高级别肿瘤相较于低级别肿瘤体积较大;体素体积(original shape voxel volume)是指在ROI 中通过体素数量与单个体素体积的相乘来计算ROI体积,体素体积精确性欠佳但可以用来评价ROI 大致体积,结果同样显示高级别肿瘤具备较大的体积;表面积(original shape surface area)直观的评价ROI 表面积,在高级别组中特征值越大则说明肿瘤原发灶表面积越大。同样在三维视角下,相较于低级别肿瘤,处于高级别肿瘤阶段的肿瘤表面积和肿瘤体积也更大(图3)。已有研究表明,肿瘤体积和表面积是影响CCRCC 患者预后的独立因素,并且WHO/ISUP 分级较高的患者预后更差[8]。
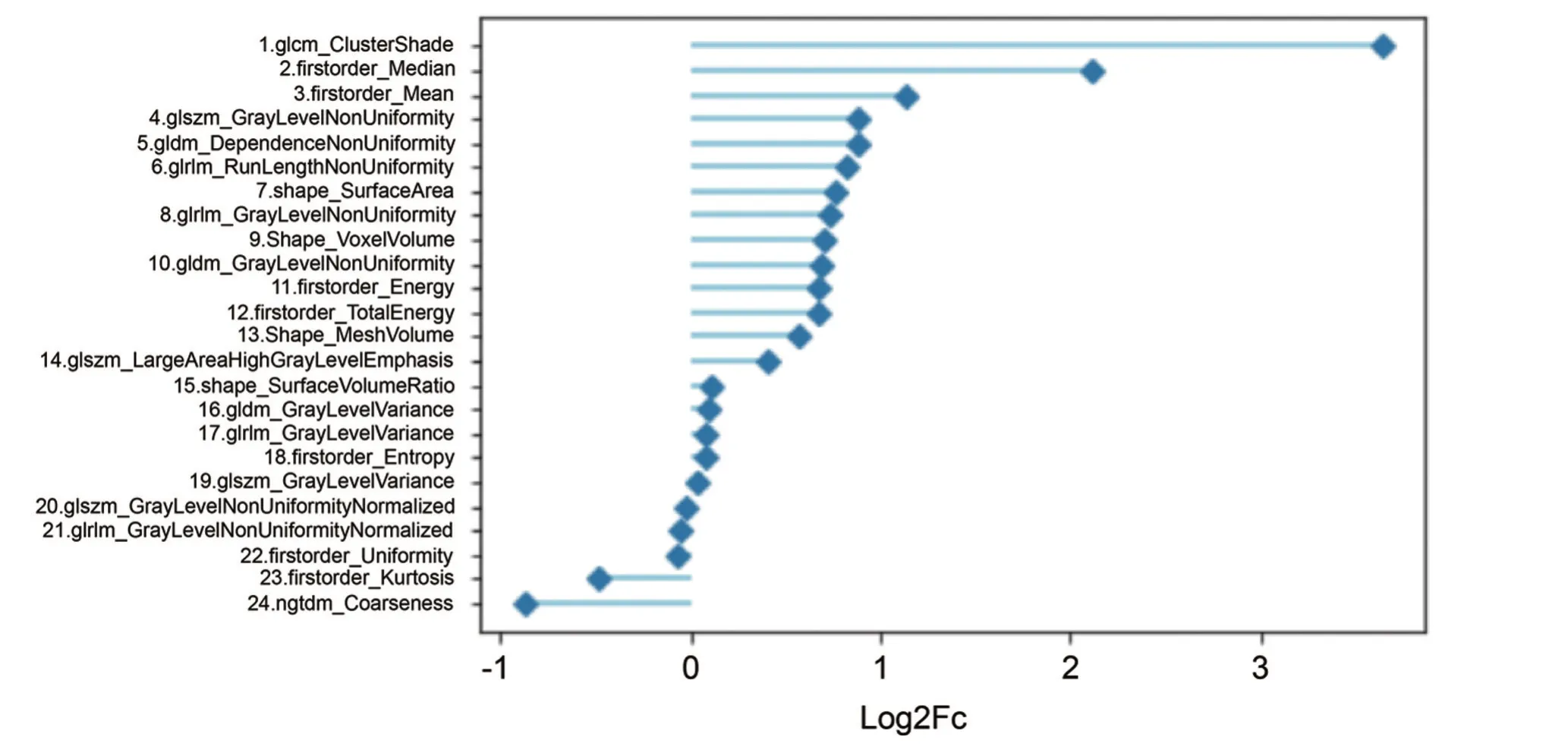
图1 与CCRCC不同风险分级显著相关的影像组学特征Figure 1 Imaging features significantly associated with different risk grades of CCRCC

图2 影像组学特征在不同肿瘤分级中的特征值Figure 2 Feature values of radiomics features in different tumor grades
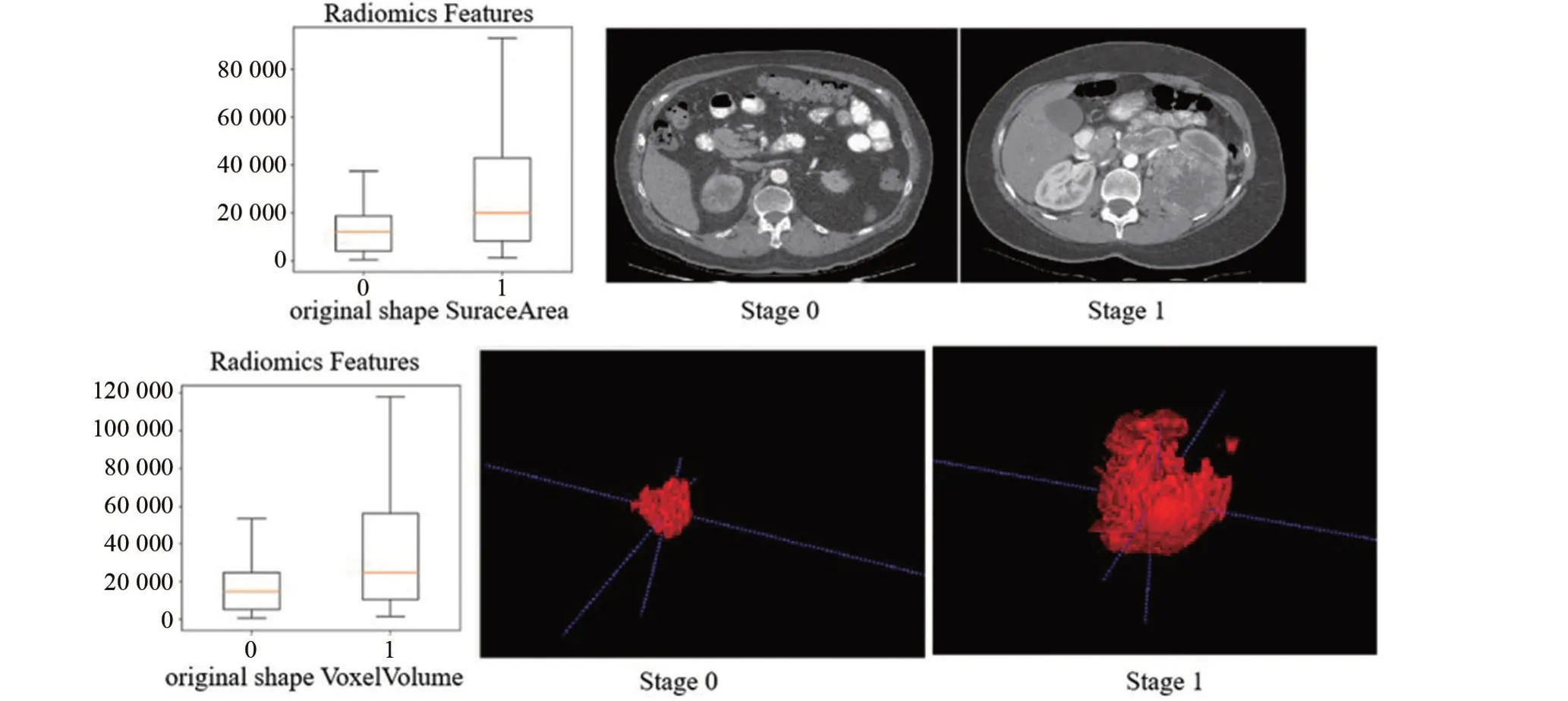
图3 三维视角下肿瘤表面积及肿瘤体积Figure 3 Tumor surface area and tumor volume in three-dimensional view
2.2 识别与CCRCC相关的基因组学特征 接下来通过Wilcoxon秩和检验,识别了在CCRCC不同肿瘤分级中表现出显著差异表达的2 125 个基因[8](图4)。此外,通过Fisher精确检验,筛选出了在肿瘤分级中其突变具有显著差异的37个基因,其中BAP1和SETD2在高级别CCRCC 患者中具有较高的突变比例(图5),并且已有研究证实了,BAP1和SETD2突变在CCRCC疾病具备病理意义,并可能引起较差的临床结局[9-10]。

图4 在CCRCC不同肿瘤分级中存在显著差异的表达基因Figure 4 Expressed genes with significant differences in different tumor grades of renal clear cell carcinoma

图5 BAP1和SETD2在高级别CCRCC中突变占比较高Figure 5BAP1 and SETD2 have a high mutation ratio in high-grade CCRCC
2.3 机器学习模型的建立与评价 结合先前识别的影像组学特征和多组学特征,进一步通过LASSO降维得到整合的关键特征。随后基于四种机器学习算法分别构建预测模型,以评估影像基因组学联合特征针对CCRCC分级的预测效能。所有模型训练过程中训练集均占比80%,测试集占比20%。为避免因数据集随机划分时导致的过拟合,在模型构建过程中本研究设定了五折交叉验证。经不同机器学习算法拟合后,绘制受试者工作特征曲线(receiver operating characteristic curve,ROC),并通过曲线下面积(area under curve,AUC)评价模型的预测效能。结果表明,使用联合特征基于SVM 算法构建的预测模型与其他模型相比具有最好的预测效能,其训练集AUC 为0.999 (95%CI:0.994~1.004),测试集AUC为0.929(95%CI:0.841~1.018)(图6)。其次是基于LR算法构建的预测模型,其训练集AUC为1.0(95%CI:1.0~1.0),测试集AUC为0.894(95%CI:0.787~0.100 1)。尽管基于RF 算法和XGB 算法构建的预测模型其预测效能最差,但是仍然可以较好的区分CCRCC的肿瘤分级(AUC>0.8),见图6。
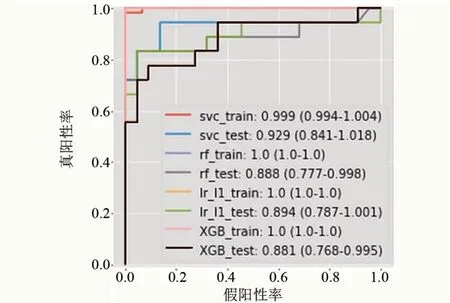
图6 基于四种机器学习模型ROC曲线Figure 6 ROC curve based on four machine learning models
上述结果表明基于影像基因组学联合特征构建的SVM 模型在预测CCRCC分级中表现最佳,因此随后使用SVM 算法,基于影像组学特征、转录组特征和基因组特征,分别构建机器学习模型,以评估不同组学识别的关键特征在CCRCC 肿瘤分级方面的预测效能(图7)。其中影像组学预测模型其AUC 为0.715(95%CI:55.1%~87.8%),转录组预测模型AUC为0.856(95%CI:73.2%~98%),基因组预测模型AUC 为0.652(95%CI:47.8%~82.5%)。结果表明,与其他水平的特征相比,基于转录组水平关键特征基因构建的SVM模型在肿瘤分级方面具有更优的预测效能(AUC=0.856,95%CI:73.2%~98%)。而结合多组学特征的影像基因组学模型(AUC=0.929,95%CI:84.1%~100%)较单一组学(AUC=0.652~0.856)模型预测效能更佳。
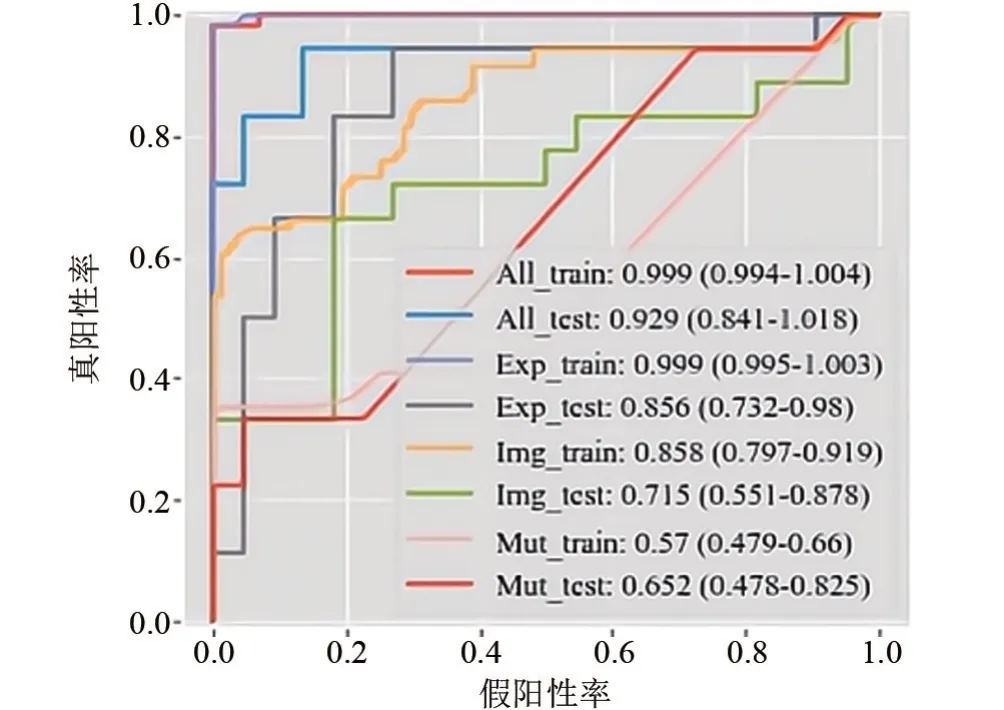
图7 基于支持向量机的单一组学模型评价图Figure 7 Model evaluation diagram of support vector machine based on single omics
2.4 CCRCC相关基因组学的特征功能 为了进一步挖掘特征基因在CCRCC 肿瘤进展中发挥的作用,本研究使用LASSO算法进一步筛选在肿瘤分级中显著差异表达的关键特征基因进行功能富集分析,以探究其参与调控的通路与功能。关键基因被富集在了多条条通路中,并按照P值排序选取了前8 条通路进行可视化展示(图8)。本研究发现与肿瘤分级相关的特征基因主要富集在了T细胞分化(T cell differentiation)、淋巴细胞分化(Lymphocyte differentiation)、细胞基质黏附(regulation of cell-matrix adhesion)和非典型Wnt 信号通路(Non-canonical Wnt signaling pathway)等生物学过程中。随后对生物学过程进行GO语义相似性聚类,结果表明特征基因主要富集在免疫相关细胞分化、细胞基质调节和细胞周期负调控等相关功能上(图9)。为进一步可视化特征基因与生物学过程之间的关系,本研究绘制了基因-功能网络图,结果显示WNT4 处于网络图的中心,相比于LBX2 和MCM9 等基因,WNT4 显然参与了更多的生物学过程(图10),并且在高级别肿瘤中WNT4 表达较高(图11),WNT4 可能在CCRCC 进展中发挥更重要的作用。
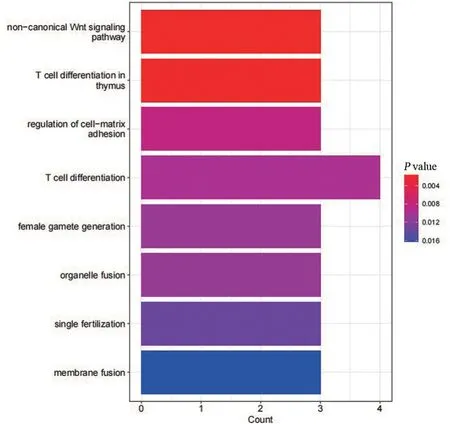
图8 基因功能富集分析Figure 8 Gene functional enrichment analysis
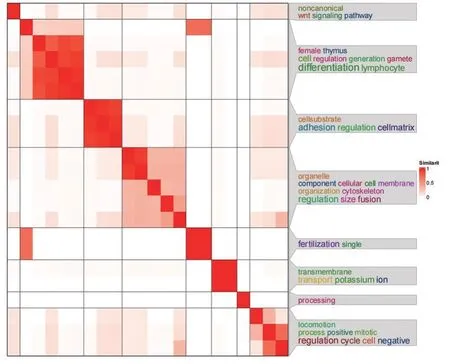
图9 功能相似性聚类Figure 9 Functional similarity clustering
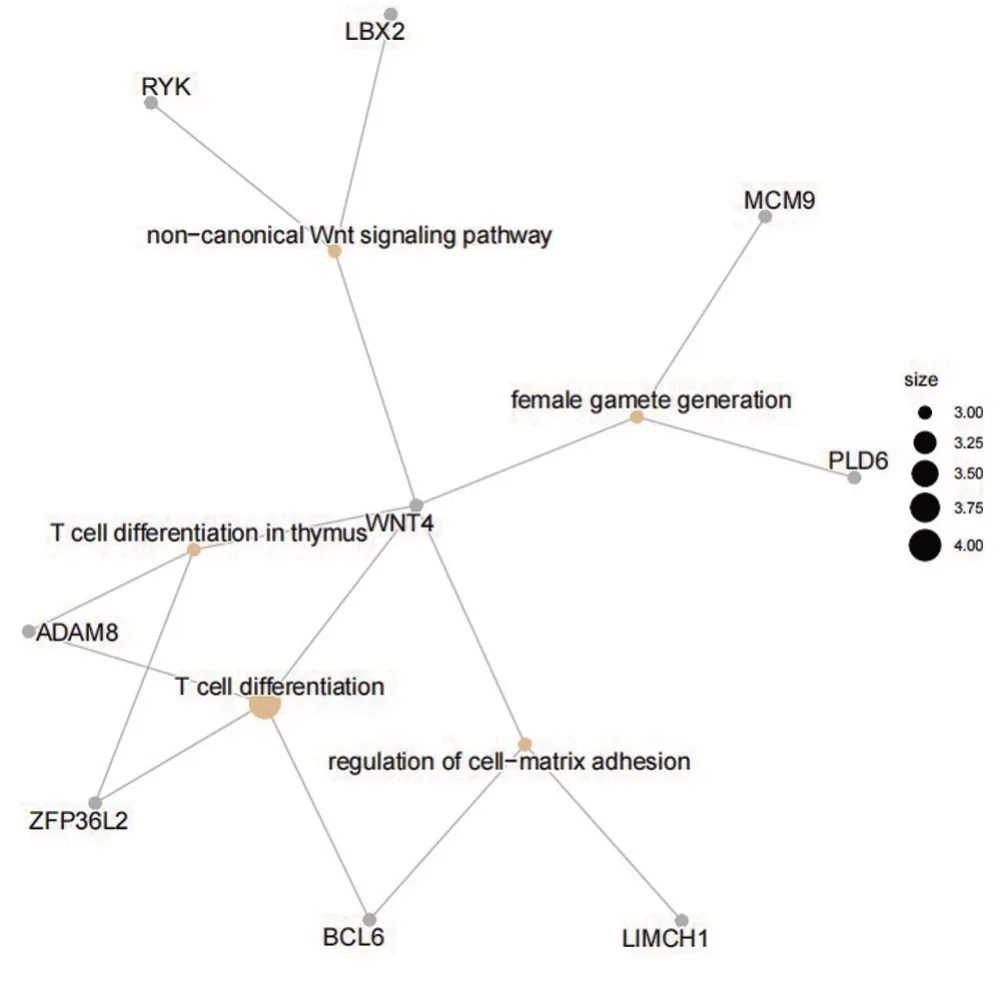
图10 基因功能网络图Figure 10 Gene function network diagram
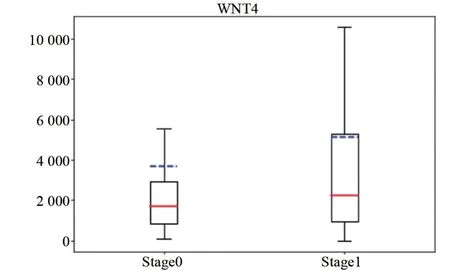
图11 WNT4在不同肿瘤分级中的表达Figure 11 WNT4 expression in different tumor grades
3 讨论
影像基因组学将患者影像资料和基因组数据相关联,广泛应用在肿瘤诊疗过程中[11-13]。本研究应用影像基因组学联合特征构建了预测CCRCC肿瘤分级模型,对比四种机器学习算法在预测CCRCC 肿瘤分级效能的不同。SVM在预测CCRCC肿瘤分级中表现最佳,AUC为0.929(95%CI:0.841~1.018)。RF、LR及XGB 模型同样能较准确预测CCRCC 肿瘤的分级(AUC=0.881~0.894)。国外同样也有研究发现SVM较K近邻模型、RF模型更能反应肺癌患者的预后[14]。其可能原因为SVM结构简单、适应性好及泛化能力强等优势和二分类问题更契合有关[15]。因此本研究利用SVM 分别构建预测CCRCC 肿瘤分级的影像组学、基因组及转录组的单组学模型,并与联合模型进行对比。结果显示影像组学预测模型AUC为0.71(95%CI:0.551~0.878),转录组预测模型AUC为0.856(95%CI:0.732~0.98),基因组预测模型AUC 为0.65 (95%CI:0.478~0.825)。这表明转录组模型在单组学预测模型中表现更优,可能与差异表达的特征基因数量较多有关。同时,联合模型中输入特征数量及特征维度较多,本实验研究结果指出联合组学模型(AUC=0.929)较单一组学(AUC=0.652~0.856)模型可更准确预测肿瘤分级。之前的研究也证实了联合模型的效能更佳[14,16]。本研究为CCRCC肿瘤分级诊断构建的影像基因组学预测模型具有临床实用性,其中基于SVM算法的多组学预测模型表现最佳,为个体化无创性肿瘤分级提供了支持工具,辅助临床术前决策。
本实验应用TCGA 数据库中多组学数据,通过Wilcoxon 秩和检验及Fisher 精确检验筛选出CCRCC肿瘤不同分级间存在显著差异表达和差异突变的基因。通过功能富集分析发现,WNT4 表达基因在功能网络中处在“枢纽”的位置(图10),能够调节非典型WNT信号通路、T细胞分化及胸腺T细胞分化[17]、雌配子传代和细胞基质的黏附。根据大多数临床研究发现CCRCC 男性发病率高于女性,结合本研究结果表明可能与WNT4 参与调控雌配子传代相关[18-19]。同时,WNT4 参与调控细胞分化会引起CCRCC 的不良预后[20],与本研究结果中WNT4 在高级组中表达较高相符[21]。本研究探索了与CCRCC相关的关键基因,揭示了关键基因参与调控的通路及功能,为CCRCC 的精准治疗提供了潜在生物学标志以及新的治疗可能。
本研究同样存在局限性。为了拓展特征维度采用多组学联合数据,符合纳入标准的样本量仅为197例,在未来的研究中可扩增数据容量,避免选择性偏差。本研究联合影像组学数据与基因组学数据构建预测模型,增加了特征维度,并且研究结果较目前国内外大多数研究具有更优的预测效能。同时,结合机器学习算法及功能富富集分析为CCRCC患者提供了无创的分级诊断方式,并为靶向治疗提供了潜在的生物学标志。
