伴有早发性高度近视的遗传性眼病基因型与表型研究
2023-08-14芮雪任英华杨尚英程婉玉容维宁盛迅伦
芮雪 任英华 杨尚英 程婉玉 容维宁 盛迅伦
1甘肃爱尔眼视光医院,兰州 730050;2宁夏回族自治区人民医院眼科 宁夏眼科医院 宁夏致盲性眼病临床医学研究中心,银川 750002
近视在全球范围内日益流行,特别是在东亚地区[1-3]。根据世界卫生组织报告,未矫正近视是全球视力损害的主要原因[4]。屈光度≤-6.00 DS列为高度近视,高度近视多伴有严重的眼部并发症,如黄斑变性、视网膜脱离、白内障和青光眼,是不可逆盲的主要原因[1-5]。高度近视分为发生在学龄前(<7岁)的早发性高度近视(early-onset high myopia,eoHM)和发生在学龄后的迟发性高度近视(late-onset high myopia,loHM)[6]。虽然近视是环境因素和遗传因素共同作用的结果,但尚无一个理论能完整地阐述近视的病因。eoHM患儿面临的环境压力风险较少,因此,其病因多由遗传倾向引起。eoHM是符合孟德尔遗传规律的单基因遗传病,遗传方式有常染色体显性(autosomal dominant,AD)、常染色体隐性(autosomal recessive,AR)和X染色体连锁(X-linked,XL)[7]。迄今为止,通过全外显子组测序和单基因连锁分析,共发现10个与eoHM相关的基因突变,其中5个与常染色体显性eoHM相关,3个与常染色体隐性eoHM相关,2个与X-连锁eoHM相关。约10%的eoHM患者(eoHM是唯一的临床特征,无眼部或全身异常)携带这些基因突变。此外,在仅约23.8%的eoHM先证者中可检测到RetNet(https://sph.uth.edu/retnet/)中列出的基因突变。其余67%~70%eoHM患者的遗传因素尚未明确[10]。我们在临床工作中发现,一些遗传性视网膜疾病和综合征,如家族性渗出性玻璃体视网膜病变(familial exudative vitreoretinopathy,FEVR)、先天性静止性夜盲(congenital stationary night blindness,CSNB)、全色盲、X-连锁遗传视网膜色素变性(X-linked retinitis pigmentosa,XLRP)、Leber先天性黑矇(Leber congenital amaurosis,LCA)、回旋状脉络膜视网膜萎缩(gyrate atrophy of the choroid and retina,GA)和Stickler综合征等,往往伴有eoHM。由于眼科医生对这一类遗传性眼病的认识与重视程度不够,易导致患者被误诊或漏诊。本研究通过高通量测序技术检测伴有eoHM的遗传性眼病患者的致病基因突变,结合患者临床表现,分析基因型及其与表型的关系,并检索文献回顾分析既往已报道的伴有eoHM的遗传性眼病研究数据,旨在帮助眼科医生更全面地了解eoHM遗传因素及可能存在的潜在疾病,提高临床诊断水平。
1 资料与方法
1.1 一般资料
采用家系调查研究方法,纳入2019年1月至2020年6月于宁夏眼科医院首诊诊断为eoHM的家系20个,家系中的先证者发病年龄为4~7岁,屈光状态≤-6.00 DS,其中男12例,女8例;汉族14例,回族6例。纳入标椎:通过患者本人或家属代述患者在学龄前(<7岁)于本院或外院行相关检查诊断为高度近视[球镜度≤-6.00 D或眼轴长度>26 mm];除眼部症状外不排除有全身系统临床体征。本研究遵循《赫尔辛基宣言》,研究方案经宁夏回族自治区人民医院伦理委员会审核批准(批文号:2016018)。所有纳入的研究对象或未成年患儿监护人均被告知本研究目的及流程,并签署遗传性眼病知情同意书。
1.2 方法
1.2.1眼科检查和病史收集 完善先证者及其家庭成员的相关眼科检查,包括裂隙灯显微镜、间接检眼镜、裸眼视力(uncorrected visual acuity,UCVA)、最佳矫正视力(best corrected visual acuity,BCVA)、视野(750i,美国Carl Zeiss Meditec公司)、色觉(色盲检查图第五版)、彩色眼底照相(TRC-NW300,日本Topcon公司)、频域光学相干断层扫描(optical coherence tomography,OCT)(HD-OCT4000,美国Carl Zeiss Meditec公司)、荧光素眼底血管造影(fluorescein fundus angiography,FFA)、眼底自发荧光、光学相干断层扫描血管成像(optical coherence tomography angiography,OCTA)及视网膜电图(electroretinogram,ERG)检查。详细询问并记录先证者及其家庭成员现病史、既往史、个人史、家族史及婚育史,绘制家系图。
1.2.2全基因组DNA提取及基因变异筛选 采集受试者外周静脉血5 ml,采用Qiamp Blood Mini Kit DNA提取试剂盒(德国Qiagen公司)提取基因组DNA。本研究所采用的目标序列捕获芯片包括232个由RetNet网站公布的遗传性视网膜疾病已知致病基因,针对已知致病基因的外显子区域、相邻内含子区域(50 bp)及已知内含子突变,采用高通量测序仪(Illumina HiSeq TM2000平台,美国Illumina公司)进行测序。平均测序深度>100 X,测序深度≥30 X的覆盖度大于98%(上海韦翰斯生物医药科技有限公司)。采用Burrows Wheeler Aligner软件(BWA version 0.6.1,http://bio-bwa.sourceforge.net/)将高通量二代测序结果与UCSC人类基因组对照序列hg19人类参考基因组序列(http://genome.ucsc.edu/index.html)进行比对和鉴别遗传变异,应用AtlasIndel2和AtlasSNP软件进行小分子的插入或缺失变异和单核苷酸变异分析。对于罕见的遗传性疾病,测序结果中所检测到的突变频率高于0.5%(隐性变异)或0.1%(显性变异),则被否认是致病突变从而被过滤。将检测获得的DNA序列变异信息与单核苷酸多态性数据库进行比对过滤,包括NIEHS外显子测序数据库、NHLBI外显子测序数据库、内部数据库、dbSNP135和千人基因组(1000 Genomes)。将高频率的变异过滤掉后,采用ANNOVAR软件对剩余的突变基因进行蛋白质的改变预测,同时剔除同义突变。
1.2.3基因变异致病性分析 对发现的可疑基因变异,在人类基因突变数据库(HGMD:http://www.hgmd.cf.ac.uk)中查看是否为已报道致病突变。对于未报道过的新变异,依据美国医学遗传学与基因组学学会2015年发布的《序列变异解读标准和指南》进行基因变异致病性评估。采用Polyphen-2 (http://genetics.bwh.harvard.edu/pph2/)、MutationTaster (http://mutationtaster.org/)、SIFT (http://sift.jcvi.Org/www/SIFT_chr_coords_submit.html)、PMut (http://mmb2.pcb.ub.es:8080/PMut/)、CADD (https://cadd.gs.washington.edu/snv)和GERP++ (http://wannovar.wglab.org/)进行致病性预测。查看1000Genome (http://browser.1000genomes.org/index.html)、EVS (http://evs.gs.washington.edu/EVS/)和ExAC (http://exac.broadinstitute.org/)数据库中变异在正常人群中的等位基因频率。利用Sanger测序在患者家属中进行共分离分析。
1.2.4文献回顾 在PubMed上以“Early onset high myopia”、“RetNet genes”、“Mutation”为检索词检索2000年1月至2020年12月相关文献,文献纳入标准:研究类型包括综述及临床研究;研究对象发病年龄在10岁之前(含10岁),且均诊断为单眼或双眼高度近视,可伴有全身系统疾病。在中国知网、万方数据及维普中文数据库上以“家族性渗出性玻璃体视网膜病变”、“X-连锁遗传视网膜色素变性”、“先天性静止性夜盲”、“Stickler综合征”、“全色盲”、“Leber先天性黑矇”、“回旋状脉络膜视网膜萎缩”为检索词检索2000年1月至2020年12月相关文献,文献纳入标准:(1)研究类型为临床研究,排除会议摘要;(2)研究方法与本研究相似;(3)临床资料完整,支持基因结果并且有疾病具体诊断标准。
2 结果
2.1 临床表型及基因检测
纳入的20个eoHM家系中,先证者均为高度近视,先证者父母双眼均为轻度近视或无近视表现,其中8个家系检测到遗传性眼病相关致病基因变异,临床表现见表1,基因检测结果见表2,其余12个家系未检测到与眼部或综合征相关的致病基因。
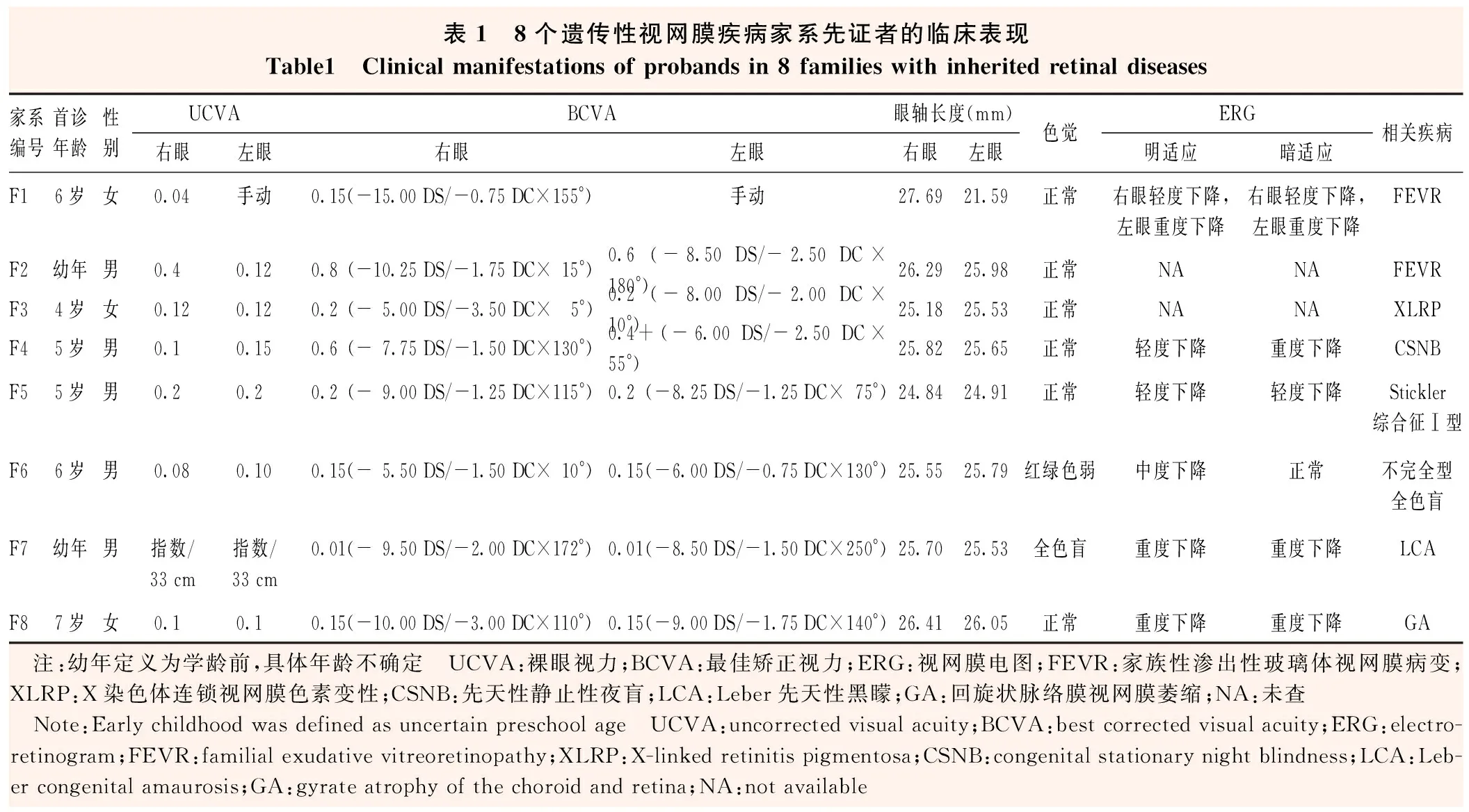
表1 8个遗传性视网膜疾病家系先证者的临床表现Table1 Clinical manifestations of probands in 8 families with inherited retinal diseases家系编号首诊年龄性别UCVABCVA眼轴长度(mm)右眼左眼右眼左眼右眼左眼色觉ERG明适应暗适应相关疾病F16岁女0.04手动0.15(-15.00 DS/-0.75 DC×155°)手动27.6921.59正常右眼轻度下降,左眼重度下降右眼轻度下降,左眼重度下降FEVRF2幼年男0.40.120.8(-10.25 DS/-1.75 DC×15°)0.6(-8.50 DS/-2.50 DC×180°)26.2925.98正常NANAFEVRF34岁女0.120.120.2(-5.00 DS/-3.50 DC×5°)0.2(-8.00 DS/-2.00 DC×10°)25.1825.53正常NANAXLRPF45岁男0.10.150.6(-7.75 DS/-1.50 DC×130°)0.4+(-6.00 DS/-2.50 DC×55°)25.8225.65正常轻度下降重度下降CSNBF55岁男0.20.20.2(-9.00 DS/-1.25 DC×115°)0.2(-8.25 DS/-1.25 DC×75°)24.8424.91正常轻度下降轻度下降Stickler综合征Ⅰ型F66岁男0.080.100.15(-5.50 DS/-1.50 DC×10°)0.15(-6.00 DS/-0.75 DC×130°)25.5525.79红绿色弱中度下降正常不完全型全色盲F7幼年男指数/33 cm指数/33 cm0.01(-9.50 DS/-2.00 DC×172°)0.01(-8.50 DS/-1.50 DC×250°)25.7025.53全色盲重度下降重度下降LCAF87岁女0.10.10.15(-10.00 DS/-3.00 DC×110°)0.15(-9.00 DS/-1.75 DC×140°)26.4126.05正常重度下降重度下降GA 注:幼年定义为学龄前,具体年龄不确定 UCVA:裸眼视力;BCVA:最佳矫正视力;ERG:视网膜电图;FEVR:家族性渗出性玻璃体视网膜病变;XLRP:X染色体连锁视网膜色素变性;CSNB:先天性静止性夜盲;LCA:Leber先天性黑矇;GA:回旋状脉络膜视网膜萎缩;NA:未查 Note:Early childhood was defined as uncertain preschool age UCVA:uncorrected visual acuity;BCVA:best corrected visual acuity;ERG:electro-retinogram;FEVR:familial exudative vitreoretinopathy;XLRP:X-linked retinitis pigmentosa;CSNB:congenital stationary night blindness;LCA:Leb-er congenital amaurosis;GA:gyrate atrophy of the choroid and retina;NA:not available

表2 8个遗传性视网膜疾病家系先证者的基因检测Table 2 Genetic test results of probands in 8 families with inherited retinal diseases家系编号染色体核苷酸改变蛋白质改变变异基因突变类型遗传方式F1chr 11c.A313Gp.Met105ValFZD4错义突变ADF2chr 7c.14_15insAAGAp.Asp5fs*TSPAN12移码突变ADF3chr Xc.2234_2237delp.Arg745fs*RPGR移码突变XLDF4chr 4c.C481Tc.C355Tp.Gln161Ter*p.Arg119Cys*GPR179移码突变ARF5chr 12c.1659_1660insACGGTGACCCTGGCCGTCCTGGp.Pro554fs*COL2A1移码突变*-F6chr 4c.C181Tc.G967Ap.Thr604Ile*p.Gly323SerPDE6B错义突变ARF7chr 17c.604_619delTCCACGGCACTCAGGGc.995G>Cp.Ser202fs*p.Arg332ProGUCY2D移码突变错义突变ARF8chr 10c.C722Tp.Pro241LeuOAT错义突变AR 注:*:新发现的基因突变;AD:常染色体显性遗传;XLD:X染色体连锁显性遗传;AR:常染色体隐性遗传;-:未知 Note:*:novel gene mutation;AD:autosomal dominant inheritance;XLD:X-linked dominant inheritance;AR:autosomal recessive inheritance;-:unknown
2.1.1FEVR家系的基因型与表型 F1先证者,女,6岁,因双眼视力低下就诊于宁夏眼科医院。眼科检查:BCVA右眼0.2(-9.25 DS/-1.25 DC×161°),左眼0.4(-7.00 DS/-0.75 DC×94°);10年后因双眼视力下降明显复查,检查发现BCVA右眼0.15(-15.00 DS/-0.75 DC×155°),左眼手动,左眼视网膜脱离;FFA呈典型FEVR改变(图1)。基因检测先证者及其父亲FZD4基因杂合性错义变异c.313A>G(p.Met105Val)(图2)。先证者父亲双眼低度近视,FFA提示周边视网膜血管分支增多,末端血管异常吻合。

图1 F1家系先证者彩色眼底照相及FFA图像 A:右眼彩色眼底照相 眼底豹纹状改变,后极部视网膜血管纤直,向颞侧牵拉(箭头) B,C:右眼和左眼FFA图像 双眼视网膜血管未发育到周边,无血管区和正常的血管化形成明显的嵴样分界,周边视网膜血管分支增多,末端血管异常吻合,有少量渗漏(箭头)Figure 1 Color fundus photographs and FFA images of proband of F1 A:Color fundus photograph of the right eye Tessellated fundus,straight retinal vessels in the posterior pole pulling toward the temporal side (arrow) were seen B,C:FFA images of right and left eyes The retinal vessels did not develop to the periphery,and the avascular area and the normal vascularization formed a clear crista-like boundary.The peripheral retinal vascular branches increased,and abnormal terminal vessel anastomosis with a little leakage (arrow) were seen

图2 F1家系图及基因测序图 A:F1家系图 B:F1家系FZD4基因测序图 Ⅰ1和先证者(Ⅱ1)均携带FZD4基因M:c.313A>G(p.Met105Val)杂合错义变异 ○:正常女性;●:患病女性;■:患病男性;:先证者Figure 2 Family pedigree and gene sequencing map of proband of F1 A:Family pedigree of proband F1 B:Location of variant in the FZD4 gene The proband (Ⅱ1) and her father (Ⅰ1) both carried a heterozygous missense variant M:c.313A>G (p.Met105Val) of FZD4 gene ○:normal female;●:female patient;■:male patient;:proband
F2先证者,男,18岁,自幼视物不清,欲行屈光矫正手术就诊于宁夏眼科医院。既往史:6岁诊断为双眼高度近视,左眼屈光性弱视,并持续接受戴镜+弱视训练治疗。BCVA右眼为0.8(-10.25 DS/-1.75 DC×15°),左眼为0.6(-8.50 DS/-2.50 DC×180°),眼底照相及FFA提示双眼视网膜血管走形异常,周边视网膜血管末端异常吻合、渗漏(图3)。基因检测先证者及其姐姐和母亲TSPAN12基因杂合性移码变异c.14_15insAAGA(p.Asp5fs*)(图4)。先证者母亲表型正常,姐姐低度近视,FFA检查提示周边视网膜血管分支繁多,呈毛刷状改变。

图3 F2家系先证者彩色眼底照相及FFA图像 A、B:右眼和左眼彩色眼底照相 右眼后极部血管走形异常(箭头);左眼后极部血管纤直(箭头),可见黄斑颞侧灰白色病灶,两者尖端相对,形成“V”形区域(*) C~F:双眼FFA图像 双眼视网膜周边血管分支增多,血管末端部分动静脉异常吻合(箭头),新生血管生成,可见渗出病灶(*)(C、D:右眼;E、F:左眼)Figure 3 Color fundus photographs and FFA images of proband of F2 A,B:Color fundus photographs of right and left eyes In the posterior pole,the blood vessels arrangement was abnormal (arrow) in the right eye,and the blood vessels were straight (arrow) in the left eye.Gray and white lesions of the temporal macula were visible in the left eye,and the tips of the two lesions were opposite to each other,forming a V-shaped area (*) C-F:Binocular FFA images Peripheral retinal blood vessels increased,and abnormal arteriovenous anastomosis at the end of blood vessels (arrow),neovascularization and exudative lesions were visible (*)(C-D:right eyes;E-F:left eyes)

图4 F2家系图及基因测序图 A:F2家系图 B:F2家系TSPAN12基因测序图 Ⅰ2、Ⅱ1和先证者(Ⅱ2)均携带TSPAN12基因M:c.14_15insAAGA(p.Asp5fs*)杂合移码变异 ○:正常女性;□:正常男性;●:患病女性;■:患病男性;:先证者Figure 4 Family pedigree and gene sequencing map of proband of F2 A:Family pedigree of proband F2 B:Location of variant in the TSPAN12 gene The proband (Ⅱ2),her mother (Ⅰ2) and her sister (Ⅱ1) all carried a heterozygous frameshift variant M:c.14_15insAAGA (p.Asp5fs*) of TSPAN12 gene ○:normal female;□:normal male;●:female patient;■:male patient;:proband
2.1.2XLRP患者的基因型与表型 F3先证者,女,4岁,因双眼视力差就诊于宁夏眼科医院。眼科检查:BCVA右眼0.2(-5.00 DS/-3.50 DC×5°),左眼0.2(-8.00 DS/-2.00 DC×10°);双眼眼前节未见明显异常,眼底未见明显异常。先证者父亲(Ⅰ1)自幼夜视力较差,双眼眼前节未见明显异常,眼底检查显示双眼眼底豹纹状改变,视盘蜡黄,血管纤细,周围视网膜可见大量骨细胞样色素沉着(图5)。基因检测先证者(Ⅱ1)携带RPGR基因杂合性变异,先证者父亲(Ⅰ1)RPGR基因半合子移码变异c.2234_2237del(p.Arg745fs*)(图6)。

图5 F3家系患者彩色眼底照相及OCT图 A、B:Ⅰ1患者右眼和左眼广角彩色眼底照相 双眼眼底豹纹状改变,视盘蜡黄,血管纤细,周围视网膜可见大量骨细胞样色素沉着 C、D:Ⅰ1患者右眼和左眼黄斑OCT 双眼黄斑水肿,结构不清,黄斑区萎缩 E、F:Ⅱ1患者右眼和左眼彩色眼底照相 双眼未见明显异常Figure 5 Color fundus photographs and OCT images of patient of F3 A,B:Wide-angle fundus photographs of right and left eyes in Ⅰ1 Tessellated fundus,yellow optic disc,thin blood vessels,and a large amount of bone-like pigmentation in the surrounding retina were seen in both eyes C,D:Macular OCT images of right and left eyes in Ⅰ1 Macular edema with unclear structure and macular atrophy were seen E,F:Color fundus photographs of right and left eyes in Ⅱ1 No obvious abnormality was seen

图6 F3家系图及基因测序图 A:家系图 B:RPGR基因测序图 Ⅰ1携带RPGR基因半合子移码变异M:c.2234_2237del(p.Arg745fs*);Ⅱ1携带RPGR基因杂合性变异 ○:正常女性;■:患病男性;:女性携带者;:先证者Figure 6 Family pedigree and gene sequencing map of F3 A:Family pedigree B:Location of variants in the RPGR gene Ⅰ1 carried a hemizygous frameshift variant M:c.2234_2237del (p.Arg745fs*) of RPGR gene,and Ⅱ1 carried a heterozygous variant of RPGR gene ○:normal female;■:male patient;:female carrier;:proband
2.1.3CSNB患者的基因型与表型 F4先证者,男,31岁,5岁时夜盲,双眼高度近视,无进行性加重。眼科检查:UCVA右眼0.1,左眼0.15;BCVA右眼:0.6(-7.750 DS/-1.50 DC×130°),左眼0.4+(-6.00 DS/-2.50 DC×55°);裂隙灯显微镜检查双眼眼前节未见明显异常,双眼眼底豹纹状改变,视盘颞侧可见脉络膜萎缩弧;OCT检查显示黄斑区厚度正常,结构清晰,椭圆体带消失(图7);色觉正常。基因检测先证者GPR179基因复合杂合性突变c.481C>T(p.Gln161Ter)和c.355>T(p.Arg119Cys*)(图8),文献数据库未有突变位点c.355>T(p.Arg119Cys*)的相关报道,ClinVar数据库无该位点致病性分析结果;生物信息学蛋白功能综合性预测软件MutationTaster预测为致病突变。

图7 F4家系先证者彩色眼底照相和OCT图 A、B:右眼和左眼彩色眼底照相 眼底豹纹状改变,视盘颞侧可见脉络膜萎缩弧 C、D:右眼和左眼黄斑OCT图 黄斑区厚度正常,结构清晰,椭圆体带消失Figure 7 Color fundus photographs and OCT images of proband of F4 A,B:Color fundus photographs of right and left eyes Tessellated fundus and atrophic arc of choroid on the temporal side of the optic disc were seen in both eyes C,D:Macular OCT images of right and left eyes The thickness of the macular area was normal,and the structure was clear,and the ellipsoid band disappeared

图8 F4家系图及基因测序图 A:家系图 B:GPR179基因测序图 先证者(Ⅰ1)携带GPR179基因复合杂合性变异,变异位点为M1:c.481C>T (p.Gln161Ter);M2:c.355>T (p.Arg119Cys*)。Ⅱ1携带M1突变 ○:正常女性;■:患病男性;:先证者Figure 8 Family pedigree and gene sequencing map of F4 A:Family pedigree B:Location of variations in the GPR179 gene The proband (Ⅰ1) carried complex heterozygous variations of GPR179 gene at M1:c.481C>T(p.Gln161Ter) and M2:c.355>T(p.Arg119Cys*).Ⅱ1 carried M1 variant ○:normal female;■:male with disease;:proband
2.1.4Stickler综合征患者的基因型与表型 F5先证者,男,5岁,因发现高度近视1年首次就诊于宁夏眼科医院。眼科检查:双眼UCVA均为0.2(欠配合);扩瞳验光右眼-9.00 DS/-1.75 DC×115°,左眼-9.00 DS/-1.75 DC×75°;BCVA右眼0.2(-9.00 DS/-1.25 DC×115°),左眼0.2(-8.25 DS/-1.25 DC×75°);裂隙灯显微镜检查双眼眼前节未见明显异常;诊断为双眼高度近视、双眼弱视。半年后复查时进一步检查发现先证者玻璃体混浊,可见带状增生膜,左眼眼底豹纹状改变,5:00位视网膜周边部可见一约1/4视盘直径大小圆形视网膜裂孔(图9);眼底自发荧光未见明显异常。详细询问先证者病史为34周早产儿,出生体质量为3 400 g,否认产伤、缺氧史,母乳喂养;无特殊家族史;儿科、骨科及耳鼻喉科辅助检查显示患者身高低于正常值(第10百分位),体质量低于正常值(第25百分位),关节活动、骨骼发育、听力均正常。基因检测发现先证者COL2A1基因上1个新发移码突变c.1659_1660insACGGTGACCCTGGCCGTCCTGG(p.Pro554fs*),其父母均未检测到该基因突变(图10)。先证者父亲表型正常,母亲双眼低度近视,左眼视网膜颞侧可见一视网膜裂孔。

图9 F5家系先证者眼前节照相、彩色眼底照相和眼底自发荧光图 A、B:右眼和左眼眼前节照相 双眼玻璃体混浊,可见较致密的膜样条带(箭头) C、D:右眼和左眼广角彩色眼底照相 右眼眼底未见明显异常,左眼眼底豹纹状改变,5:00位视网膜周边部可见一约1/4视盘直径大小的圆形视网膜裂孔(箭头) E、F:右眼和左眼眼底自发荧光图 未见明显异常Figure 9 Anterior segment photograph,color fundus photograph and autofluorescence images of proband of F5 A,B:Anterior segment photographs of right and left eyes Vitreous opacity with dense membranous strip (arrow) were seen C,D:Wide-angle fundus color photographs of right and left eyes Fundus was normal in right eye,and tessellated fundus was seen in left eye with a circular retinal hole (arrow) of about 1/4 diameter of optic disc in the periphery retina at 5:00 E,F:Fundus autofluorescence images of right and left eyes No obvious abnormalities was seen

图10 F5家系图及基因测序图 A:家系图 B:COL2A1基因测序图 Ⅰ1、Ⅰ2均未携带突变基因,Ⅱ1携带变异位点为M:COL2A1基因c.1659_1660insACGGTGACCCTGGCCGTCCTGG(p.Pro554fs*)的移码突变,该突变为新发现突变 ○:正常女性;□:正常男性;■:患病男性;:先证者 Figure 10 Family pedigree and gene sequencing map of F5 A:Family pedigree B:Location of variants in the COL2A1 gene Ⅰ1 and Ⅰ2 did not carry variants.Ⅱ1 carried a frameshift variant of M:COL2A1 gene c.1659_1660insACGGTGACCCTGGCCGTCCTGG (p.Pro554fs*),a de novo mutation ○:normal female;□:normal male;■:male patient;:proband
2.1.5不完全型全色盲患者的基因型与表型 F6先证者,男,6岁,因双眼视力低下就诊。眼科检查:UCVA右眼0.08,左眼0.10;综合验光右眼0.15(-5.50 DS/-1.50 DC×10°),左眼0.15(-6.00 DS/-0.75 DC×130°);轻度红绿色弱;双眼眼前节及眼底未见明显异常改变(图10)。无特殊家族史,先证者父母均有低度近视,余未见明显异常。基因检测先证者PDE6B基因复合杂合性突变c.1811C>T(p.Thr604Ile*)和c.967G>A(p.Gly323Ser),文献数据库未有2个位点的相关性报道,ClinVar数据库无2位点致病性分析结果;生物信息学蛋白功能综合性预测软件MutationTaster预测为致病突变;经验证先证者父亲和母亲分别携带p.Thr604Ile*及p.G323S(图11,12)。

图11 F6家系先证者广角彩色眼底照相、黄斑OCT和眼底自发荧光图 A、B:右眼和左眼广角彩色眼底照相 双眼均未见明显异常 C、D:右眼和左眼黄斑OCT 黄斑区结构清晰,未见明显异常 E、F:右眼和左眼眼底自发荧光图 未见明显异常Figure 11 Wide-angle fundus color photograph,macular OCT and fundus autofluorescence images of proband of F6 A,B:Wide-angle fundus color photographs of right and left eyes No obvious abnormalities was seen C,D:Macular OCT images of right and left eyes Macular structure was clear without no obvious abnormalities E,F:Fundus autofluorescence images of right and left eyes No obvious abnormality was seen

图12 F6家系图及基因测序图 A:家系图 B:PDE6B基因测序图 Ⅱ1在PDE6B基因上检测到复合杂合性突变M1:c.1811C>T(p.Thr604Ile*)和M2:c.967G>A(p.Gly323Ser)。Ⅰ1、Ⅰ2分别携带M1及M2各1个位点杂合突变 ○:正常女性;□:正常男性;■:患病男性;:先证者Figure 12 Family pedigree and gene sequencing map of F6 A:Family pedigree B:Location of variants in the PDE6B gene Complex heterozygous variants M1:c.1811C>T(P.Thr604Ile*) and M2:c.967G>A(p.Gly323Ser) were detected on PDE6B gene in Ⅱ1.Ⅰ1 and Ⅰ2 carried a heterozygous mutation of M1 and M2,respectively ○:normal female;□:normal male;■:male patient;:proband
2.1.6LCA患者的基因型与表型 F7先证者,男,11岁,自幼双眼畏光、高度近视、眼球震颤。眼科检查:双眼UCVA均数指/33 cm,BCVA右眼0.01(-9.50 DS/-2.00 DC×172°),左眼0.01(-8.50 DS/-1.50 DC×250°);裂隙灯显微镜检查双眼眼前节未见明显异常;广角眼底照相检查眼底出现豹纹状改变;ERG检查双眼明暗适应下,a波、b波均重度下降(图13);双眼全色盲,眼球水平震颤。先证者父亲双眼眼球水平震颤伴双眼低度近视、双眼弱视。基因检测先证者GUCY2D基因复合杂合性突变c.604_619delTCCACGGCACTCAGGG(p.Ser202fs*)和c.995G>C(p.Arg332Pro);其父亲和母亲分别携带p.Ser202fs*及p.Arg332Pro(图14)。

图13 F7家系先证者眼部外观、彩色眼底照相及ERG A、B:强光及暗光环境下眼部外观照 患者明显畏光 C、D:右眼和左眼广角眼底照相 双眼眼底豹纹状改变 E、F:双眼暗适应0.01 ERG和明适应3.0 ERG 双眼明暗适应下,a波、b波均重度下降(1为右眼,2为左眼) ERG:视网膜电图Figure 13 Ocular appearance,fundus color photograph and ERG result of proband of F7 A,B:Binocular appearance under strong light and dark light environment The proband was photophobic C,D:Wide-angle fundus photographs of right eye and left eye There were tessellated fundus changes E,F:Binocular ERG examination results of scotopic 0.01 ERG and photopic 3.0 ERG In both eyes,a and b waves decreased severely under photopic and scotopic conditions (1:right eye,2:left eye) ERG:electroretinogram

图14 F7家系图及基因测序图 A:家系图 B:GUCY2D基因测序图 先证者GUCY2D基因复合杂合性突变M1:c.604_619delTCCACGGCACTCAGGG (p.Ser202fs*);M2:c.995G>C (p.Arg332Pro) Ⅰ1、Ⅰ2分别携带M1及M2各1个位点的杂合性突变 ○:正常女性;□:正常男性;■:患病男性;:先证者Figure 14 Family pedigrees and gene sequencing map of proband of F7 A:Family pedigree B:Location of variants in the GUCY2D gene Proband (Ⅱ1) carried two complex heterozygous variations,M1:c.604_619delTCCACGGCACTCAGGG (p.Ser202fs*) and M2:c.995G>C(p.Arg332Pro)of GUCY2D gene.His parents (Ⅰ1,Ⅰ2) carried a heterozygous variant at M1 and M2,respectively ○:normal female;□:normal male;■:male patient;:proband
2.1.7GA患者的基因型与表型 F8先证者,女,20岁,7岁时发现双眼夜盲、高度近视,因近4年双眼视力明显持续下降首次就诊于宁夏眼科医院。眼科检查:BCVA右眼0.15(-10.00 DS/-3.00 DC×110°),左眼0.15(-9.00 DS/-1.75 DC×140°);裂隙灯显微镜检查双眼晶状体轻度混浊,余眼前节未见明显异常;眼底照相、FFA显示双眼视网膜脉络膜萎缩(图15)。先证者父母表型均正常。基因检测先证者位OTA基因纯合性突变c.772C>T(p.Pro241Leu);其父亲和母亲均携带同一位点的杂合性突变(图16)。

图15 F8家系先证者广角彩色眼底照相、FFA和黄斑OCTA A、B:右眼和左眼广角彩色眼底照相 双眼眼底豹纹状改变,视网膜及脉络膜萎缩,仅保留黄斑区3倍视盘直径大小,周边视网膜可见大量不规则环状、脑回状萎缩灶 C、D:右眼和左眼FFA 双眼视网膜脉络膜萎缩,可见裸露的脉络膜大血管 E、F:右眼和左眼黄斑OCTA 双眼黄斑拱环形态破坏、局部血管密度减低,黄斑水肿,未见新生血管。第1行为黄斑区视网膜层间结构和血流信号,第2行为扫描区域视网膜深层血管、浅层血管及毛细血管层的血管分布Figure 15 Wide-angle fundus color photograph,FFA and macular OCTA images of proband of F8 A,B:Wide-angle fundus color photographs of right and left eyes In both eyes,tessellated fundus,retinal and choroidal atrophy were seen with only 3 times the diameter of the optic disc preserved in macula,and a large number of visible irregular ring-shaped and cerebral gyrus-shaped atrophy foci in the peripheral retina C,D:FFA images of right and left eyes In both eyes,retinal and choroidal atrophy with large exposed choroid vessels were seen E,F:Macular OCTA images of right and left eyes In both eyes,damaged macular arch ring,decreased local blood vessel density and macular edema were seen,and no neovascularization was observed.The first line was the interlaminar structure and blood flow signal of retina in macular area.The second was the distribution of deep retinal blood vessels,superficial blood vessels and capillary layers in the scanned region

图16 F8家系图及基因测序图 A:家系图 B:OAT基因测序图 Ⅰ1、Ⅰ2携带同一基因同一位点的杂合性突变,Ⅱ1携带该基因位点的纯合性突变,变异位点为OAT基因M:c.772C>T(p.Pro241Leu) ○:正常女性;□:正常男性;●:患病女性;:先证者Figure 16 Family pedigree and gene sequencing map of F8 A:Family pedigree B:Location of variants in the OAT gene Ⅰ1 and Ⅰ2 carried heterozygous mutations at the same locus of OAT gene,and Ⅱ1 carried a homozygous variation M:c.772C>T (p.Pro241Leu) of OAT gene ○:normal female;□:normal male;●:female patient;:proband
2.2 文献数据回顾
检索相关文献,共纳入4篇文献。Marr等[8]报道的112例10岁前患有高度近视的儿童中,92%的儿童患有与高度近视相关的遗传性眼病或全身其他系统异常的综合征,其中14%为遗传性视网膜营养不良,13%为Stickler综合征或马凡综合征(表3)。Logan等[9]研究27例eoHM患者,发现44%(12/27)的患者患有与高度近视相关的遗传性眼病或全身其他系统异常的综合征(表3)。Sun等[10]对298例eoHM患者进行全外显子测序,结果显示23.8%的患者可检测到与遗传性视网膜疾病相关基因的致病性突变(表4)。Zhou等[11]对325例eoHM患者进行基因测序,发现76例(占23.4%)患者存在RetNet基因突变,共检测到33个变异基因(表4)。

表3 既往报道的儿童早期高度近视与眼部和全身疾病的关联类型及频率Table 3 Reported type and frequency of ocular and systemic diseases associated with early high myopia in childhood文献儿童早期高度近视纳入标准纳入总例数单纯性高度近视例数[n(%)]伴有相关疾病例数[n(%)]眼部疾病全身疾病相关疾病类型单纯型综合征型Marr等[8]单眼或双眼屈光度≤-6.0 D,年龄<10岁1129(8)42(38)61(54)先天性静止性夜盲、视锥细胞营养不良、眼白化病、Stargardt病Stickler综合征、马凡综合征、Ehlers-Danlo综合征、Crouzon综合征、Noonan综合征、唐氏综合征、眼皮肤白化病、WAGR综合征、Gordon综合征、Smith-Magenis综合征、Adams-Oliver综合征Logan等[9]单眼或双眼屈光度≤-5.0 D,年龄<10岁2715(56)7(25)5(19)视网膜营养不良Stickler综合征、马凡综合征、高胱氨酸尿症、先天性脊柱骨骺发育不良、Weill-Marchesani综合征

表4 既往报道的eoHM与遗传性眼病的关联类型及基因突变Table 4 Reported types and gene mutations of inherited ocular diseases associated with eoHM文献eoHM纳入标准纳入总例数RetNet基因变异例数[n(%)]伴有高度近视的遗传性眼病相关基因占比伴有高度近视的遗传性眼病相关基因相关疾病类型Sun等[10]屈光度≤-6 D或眼轴长度>26 mm,年龄<7岁29871(23.8)62.0%(44/71)COL2A1、COL11A1、PR-PH2、FBN1、GNAT1、OPA1、PAX2、GUCY2D、TSPAN12、CACNA1F、RPGRStickler综合征、视网膜色素变性、马凡综合征、家族性渗出性玻璃体视网膜病变、遗传视神经萎缩、先天性静止性夜盲Zhou等[11]屈光度≤-6 D或眼轴长度>26 mm,年龄<7岁32576(23.4)46.1%(35/76)COL2A1、COL11A1、RPGR、CACNA1F视锥细胞营养不良、先天性静止性夜盲、全色盲、视网膜色素变性 注:eoHM:早发性高度近视 Note:eoHM:early-onset high myopia
3 讨论
近年来,我们在临床工作中发现eoHM常常是遗传性眼病患者最早就诊的原因。本研究通过高通量测序技术检测伴有eoHM的遗传性眼病患者的致病基因突变,结合患者临床表现,分析其基因型及表型,旨在帮助眼科医生更全面地了解eoHM患者的遗传因素及可能存在的潜在疾病,提高遗传性眼病的早期诊断水平。而其他导致遗传性视网膜疾病以及高度近视相关综合征的突变基因可能是筛查eoHM的候选基因。因此本研究共纳入了20个先证者表现为eoHM的家系进行了研究,其中8个家系检测到致病基因,涉及7种遗传性视网膜疾病,包括FEVR、Stickler综合征、CSNB、全色盲、XLRP、LCA和GA。
FEVR是一种遗传性视网膜血管发育异常疾病,具有高度遗传异质性,基因型与表型无固定对应,多为显性遗传。多数FEVR患者在婴幼儿时期就已发病,常无早产、吸氧史,主要以高度近视、斜视就诊时被发现,部分患者病情可终生处于静止期,无明显临床症状,仅在FFA检查时发现周边视网膜血管异常[11-12]。Yang等[13]对9例FEVR患者进行回顾性研究发现,所有患者在儿童早期就已表现有近视(8岁前至少有一眼近视屈光度<-5.00 D),7例患者为弱视,由于对儿童进行周边眼底检查较困难而且轻度FEVR在缺少FFA检查时不易被发现,这些患者早期并未诊断为FEVR。本研究中的F1、F2家系的先证者均在学龄前表现为高度近视,由于缺乏周边视网膜的详细检查导致漏诊,随后造成不同程度的视力下降,甚至视网膜脱离,因此高度近视患儿能配合检查时应尽早行视网膜及造影检查,并在随访中密切观察周边视网膜的变化,在出现视网膜裂孔及渗漏时尽早治疗以挽回视力。
RP是主要以视锥细胞和视杆细胞的变性、凋亡为特征的遗传性疾病,以夜盲、进行性视野损害、ERG异常和眼底特异性改变为主要临床特征。RP的遗传方式包括AD、AR、XL遗传及其他散在遗传方式。XLRP约占所有RP家族的15%,是相对严重的RP亚型,发病早,进展快,是青少年致盲的常见原因之一。本研究团队曾报道1个6代XLRP家系,在8例男性RP患者和14例女性携带者的RPGR基因外显子ORF15检测到移码突变,其中8例男性患者均有典型的RP改变,而14例女性携带者最具特征性的临床表型是中高度近视[14]。本研究F3家系中也可见男性RP患者,女性仅表现为单纯性高度近视表型,因此在eoHM患儿就诊时需要详细询问家族史特别是亲代病史或临床表现,必要时需对家系家属进行相应检查。
CSNB是一种具有遗传性、非进展性的视网膜病变。根据视杆细胞受损情况可进一步分为2个亚型,即视杆系统无功能的完全型CNSB和视杆系统保留有一定功能的不完全型CNSB。完全型CNSB患者多表现为中度到高度近视,而不完全型CNSB患者则多表现为轻度近视或远视[15]。2种类型的CSNB眼底基本正常,完全型CSNB患者常常出现视盘倾斜、颞侧变淡,视盘边萎缩弧等高度近视眼底改变。大部分CSNB患者初诊早期主诉视物模糊,少数患者主诉夜盲。由于患者眼底正常且无夜盲主诉,在未进行ERG检查的情况下,医师很难做出正确诊断。本研究中F4家系先证者5岁时发现夜盲、高度近视,无进行性加重。就诊时眼底检查发现高度近视视网膜改变,ERG显示暗视a波、b波均重度降低,明视ERG轻度降低,结合患者基因测序结果和家系图确诊为完全型CSNB。
Stickler综合征是AD胶原结缔组织疾病(遗传性进展性关节-眼病),主要以眼部、口面部、关节及听觉损伤为特征。Stickler综合征的眼部表现包括先天性近视、玻璃体异常、白内障、青光眼和视网膜脱离;口面部改变包括腭裂、面中部扁平、低鼻梁和小颌;关节异常包括关节活动度过大和骨关节炎;听力损伤包括感觉神经性或传导性听力障碍[16-17]。Stickler综合征眼部病变尤为突出且严重,是儿童孔源性视网膜脱离常见的原因。Stickler综合征患者视网膜脱离的发生率为65%,平均年龄15.2岁,近视的发生率高于75%,且通常是6岁之前出现的先天性高度近视,近视度数稳定不进展[18-19]。目前,已发现Stickler综合征有6种亚型,其中Ⅰ型(STL1,OMIM108300)较常见,占80%~90%。STL1是一种显性遗传性疾病,由COL2A1基因突变引起(OMIM120140)。本研究中F5家系先证者首次就诊时仅表现为高度近视,未伴有其他体征,半年后复查扩瞳检查显示双眼典型的条带样玻璃体增生膜,左眼视网膜裂孔,继而怀疑遗传性眼病可能,对家系进行基因检测和验证显示携带COL2A1基因的新突变。提示在前期发现患儿未能解释的高度近视时,建议行基因检测并在随访期间密切关注眼部及全身变化。
全色盲是一种视锥细胞营养不良性疾病和严重的色觉障碍,属于完全性视锥细胞功能障碍,其临床特征包括自幼视力低下、畏光、眼球震颤、色觉完全或部分丧失。全色盲患者双眼视力差通常≤0.1且稳定。全色盲包括完全型和不完全型2种类型,其中完全型全色盲患者眼底表现大致正常,易被漏诊,仅少数患者会出现中周部视网膜色素上皮异常和视网膜血管变窄,ERG检查显示视锥细胞反应呈现熄灭型而视杆细胞反应基本正常;不完全型全色盲又称非典型全色盲,患者有部分的辨色能力,畏光和视力损害程度也较完全型全色盲轻[20]。全色盲多伴有高度近视,眼底检查正常,如未进一步做色觉及ERG等相关检查,易误诊为屈光不正性弱视。本研究中F6家系先证者首次就诊时发现双眼呈高度近视伴轻度红绿色弱,但双眼眼前节及眼底未见明显异常改变,ERG明视仅轻度降低,因此诊断为双眼弱视,戴镜校正后无法提高视力进一步行基因检测,发现携带PDE6B基因复合杂合性变异,最终诊断为全色盲。
LCA是导致婴幼儿盲的严重遗传性视网膜疾病,呈AR遗传,与出生后前几个月出现的严重视力障碍有关,伴有固视障碍、眼球震颤和畏光,常见指-眼征(患儿常用手指或手指关节按压自己的眼球)。患者早期眼底检查多正常,随着病情进展,数年后可见眼底椒盐样色素沉着、骨细胞样色素沉着、视网膜血管狭窄、广泛视网膜色素上皮和脉络膜萎缩,周边或赤道处视网膜偶见不规则、多发的黄白色点状渗出灶,也可出现视盘水肿[21]。患者ERG表现为a波、b波重度下降,甚至消失。患儿通常表现为高度远视或罕见的高度近视。本研究中F7家系先证者自幼明显畏光、视力较差,并且双眼眼球水平震颤,首次就诊于宁夏眼科医院时发现双眼高度近视,色觉为全色盲,但双眼眼前节及眼底自发荧光等检查均未见明显异常,被误诊为全色盲,1年后ERG检查发现双眼明适应及暗适应a波、b波均重度下降,结合基因检测结果最终确诊为LCA。以上2个家系均提示我们在诊断伴有eoHM的遗传性眼病时早期行视网膜电生理检查非常必要。
GA是遗传性原发性脉络膜视网膜萎缩性疾病,临床特征为眼底回旋状病损和高鸟氨酸血症。GA首发症状为夜盲,大部分GA患者10岁左右出现夜盲。由于现代人工照明的范围越来越大,早期患者很难发现夜视力差。90%的GA患者表现为-6.00 D以上高度近视和2.00 D以上高度散光[22]。由于患者早期矫正视力正常且眼底后极部多表现正常,易误诊为屈光不正性弱视。此外,无脉络膜症和GA的临床症状非常相似,其首发症状均为夜盲,且均以进行性视力损害为特征。无脉络膜症夜盲一般在10~20岁出现,随着脉络膜视网膜的进行性萎缩,视力下降呈缓慢进行性发展。GA早期的临床症状和眼底视网膜脉络膜的萎缩易被误诊为无脉络膜症。本研究中F8家系先证者7岁时发现夜盲,以夜盲为首发症状就诊于宁夏眼科医院,检查发现双眼高度近视,且扩瞳眼底检查显示双眼呈不规则视网膜脉络膜萎缩,当时误诊为无脉络膜症,13年后再次复查时可见眼底不规则环状萎缩灶的典型改变,结合基因检测结果后确诊为GA。
国内外许多研究者通过遗传学的研究方法来探索高度近视的发病机制,已经发现17个导致非综合征型单纯型高度近视的致病基因,包括ZNF644[23]、SCO2[24]、SLC39A5[25]、CCDC111[26]、P4HA2[27]、BSG[28]、CPSF1[29]、NDUFAF7[30]、TNFRSF21[31]、XYLT[32]、DZIP1[32]、LRPAP1[33]、CTSH[33]、LEPREL1[34]、LOXL3[35]、ARR3[36]和OPN1LW[37],但这些基因突变仅能解释一小部分eoHM。既往研究表明,约1/4的eoHM患者存在与遗传性视网膜疾病相关的致病性变异基因,这些致病性基因可以导致高度近视相关的遗传性眼病或全身其他系统异常的综合征,其中视网膜营养不良、Stikler综合征及马凡综合征较常见[8-11]。本研究收集到20个eoHM家系,初诊临床记录均显示为高度近视,其中8个家系检测到致病基因突变,分别为FZD4、TSPAN12、RPGR、GPR179、COL2A1、PDE6B、GUCY2D及OAT基因变异。除OAT为新发现基因外,其他7个基因与既往报道的与eoHM相关的RetNet基因一致。8个家系最终确定的疾病类型分别为FEVR Ⅰ型、XLRP、CSNB、Stickler综合征Ⅰ型、不完全型全色盲、LCA、GA。除GA外,其他6种遗传性眼病与既往报道的与eoHM相关的遗传性眼病表型一致。除F3家系的父亲(Ⅰ1)表现为典型的RP,F7家系的父亲(Ⅰ1)双眼伴有眼球震颤,其余18个eoHM家系中父母双眼为低度近视或无近视表现,未伴有其他特殊眼部或全身表型。其他12个家系先证者仅表现为单纯性高度近视,可能与环境因素有关。既往研究未对伴有eoHM的遗传性眼病患者临床资料进行分析,也未进一步分析基因型与表型的关系,本研究中对伴有eoHM的遗传性眼病的基因型和表型进行分析,证明eoHM是这些遗传性眼病最早引起患者家属及临床医生关注的体征,也为儿童高度近视遗传筛查提供了有力依据。但本研究样本量较小,未来仍需扩大样本量进一步研究。
综上所述,eoHM与一些遗传性眼病密切相关,是FEVR、XLRP、CSNB、LCA、全色盲、GA、Stickler综合征等遗传性眼病和综合征儿童最早就诊的原因及临床医生发现潜在眼部疾病的线索,进一步对eoHM患者进行特异性临床检查和遗传筛查有助于这些疾病的早期诊断和长期随访评估。建议对eoHM儿童进行眼科结构和功能的临床评估及遗传筛查,开展基于基因检测结果的遗传咨询和生育指导,以降低这些致盲眼病的发病率。未来进一步对这些突变基因的功能以及对突变携带者及其家庭成员相关表型的研究可能为探索高度近视的发病机制提供有价值的信息。
利益冲突所有作者均声明不存在利益冲突
作者贡献声明芮雪:参与试验设计、数据分析及文章撰写;任英华、杨尚英、程婉玉:直接参与研究实施、数据采集及分析;盛迅伦、容维宁:对数据分析和论文撰写进行指导
