固相萃取-气相色谱-三重四级杆串联质谱法检测生活饮用水中8种酰胺类除草剂残留
2023-08-08曹忠波谷洋张媛媛刘晓晶华正罡
曹忠波,谷洋,张媛媛,刘晓晶,华正罡
(1.辽宁省疾病预防控制中心,辽宁省环境与人群健康监测重点实验室,沈阳 110172;2.辽宁省检验检测验证中心,沈阳 110033)
酰胺类除草剂主要用来防治一年生禾本科杂草和部分阔叶杂草,对多种禾本科双子叶植物有较强的杀毒作用[1]。广泛应用于玉米、花生、大豆、棉花等农作物生产种植中,是目前农业生产中应用较为广泛的一类除草剂[2]。酰胺类除草剂的土壤吸附常数比较低,水溶性强,在农业生产中施用后易转移到地表水和浅层地下水中,对饮用水水源可能造成污染,进而威胁人类饮水安全及健康[3]。
酰胺类除草剂进入环境后,容易转化为二烷基苯醌亚胺,可增加人和动物发生癌变的风险[4-5]。甲草胺和乙草胺已先后被美国国家环境保护署(EPA)列为B-2 类致癌物[6]。近年来,酰胺类除草剂在环境样品中残留的关注度逐渐提高。于志勇等[7]采用固相萃取-气质联用检测方法调查了我国一些主要城市的水源水和出厂水中的除草剂乙草胺残留水平。结果表明,水源水中乙草胺的检出率为66.9%,乙草胺的污染比较普遍。从区域上看,东北地区的平均检出浓度远高于西北和西南地区;从水源类型上看,湖库型和河流型水源水中乙草胺浓度远高于地下型水源,污染严重。我国现行GB/T 5750—2006《生活饮用水标准检验方法》中还没有针对8种酰胺类除草剂建立检测标准。因此,建立一个快速、准确、灵敏度高检测方法,对水源水的污染状况评价及人体饮用水健康风险评估等研究有积极帮助。
目前,酰胺类除草剂农药残留的检测方法主要有气相色谱(GC)法[8-9]、气相色谱-质谱联用(GCMS)法[10]、高效液相色谱 (HPLC)法[11]、液相色谱串联质谱 (LC-MS/MS)法[12]等。气相色谱法和高效液相色谱法用于定性分析时容易出现结果假阳性、方法的检测灵敏度低等问题。串联质谱(MS/MS)法有更精准的选择性、特异性,能有效地降低噪音,提高信噪比,从而提高灵敏度和测定结果的准确度,避免假阳性或者假阴性的结果,可以很好的解决应用GC、LC、GC-MS 法检测复杂基质样品中多种酰胺类除草剂遇到的问题。
笔者采用固相萃取净化技术(SPE)对大体积水体样本进行富集、净化,并用气相色谱-三重四级杆串联质谱 (GC-MS/MS)仪进行检测,建立生活饮用水中酰胺类除草剂的快捷、高效、准确的分析方法,以满足生活饮用水中多种酰胺类除草剂残留的检测。
1 实验部分
1.1 主要仪器与试剂
气相色谱-三重四级杆质谱联用仪:GC-7890+MS/MS-7000B型 ,美国安捷伦科技有限公司。
氮吹仪:Turbo VapⅡ型,美国 Caliper公司。
超纯水机:IQ7000型,美国密理博公司。
自动固相萃取仪:Sepaths-10型,北京莱伯泰科仪器有限公司。
分析天平:XS205,感量为0.1 mg,瑞士梅特勒-托利多集团。
C18固相萃取小柱:6 mL/500 mg,美国安捷伦科技有限公司。
氨基固相萃取小柱:6 mL/500 mg,美国安捷伦科技有限公司。
HLB 固相萃取小柱:6 mL/500 mg,美国沃特世公司。
酰胺类除草剂标准样品:毒草胺(纯度不小于99.6%)、乙草胺(纯度不小于97.9%)、甲草胺(纯度不小于99.9%)、异丙甲草胺(纯度不小于98.5%)、双苯酰草胺(纯度不小于98.5%)、丁草胺(纯度不小于96.3%)、敌草胺(纯度不小于99.8%)、丙草胺(纯度不小于99.8%),德国DR.Ehrenstorfer GmbH公司。
环氧七氯B:纯度不小于99.3%,德国DR.Ehrenstorfer GmbH公司。
二氯甲烷、乙酸乙酯、丙酮、甲醇、正己烷:均为色谱纯,美国Fisher公司。
无水硫酸钠:分析纯,经450 ℃烘烤2 h,置于干燥器内备用,国药集团化学试剂有限公司。
8 种酰胺类除草剂混合标准储备溶液:质量浓度均为1.0 mg/mL,准确称取8种酰胺类除草剂标准样品各10.0 mg于10 mL容量瓶中,加入丙酮溶解并定容至标线。于4 ℃冰箱中避光保存,于6 个月内使用。
8 种酰胺类除草剂混合标准中间溶液:各组分质量浓度均为1.00 mg/L,用丙酮将8 种酰胺类除草剂混合标准储备溶液稀释、定容,置于冰箱中避光保存,于1个月内使用。
环氧七氯B 标准溶液:20.0 mg/L,准确称取环氧七氯B 10.0 mg标准样品于10 mL容量瓶中,加入丙酮溶解,定容至标线,取溶液适量,再用丙酮稀释至50 倍体积,于4 ℃冰箱中避光保存,于1 个月内使用。
氮气:纯度不小于99.999%,沈阳广泰气体有限公司。
氦气:纯度不小于99.999%,沈阳广泰气体有限公司。
实验用水:经超纯水机制得,现用现制。
1.2 仪器工作条件
1.2.1 色谱条件
色谱柱:HP-5MS UI 柱(30 m×0.25 mm,0.25 μm,美国安捷伦科技有限公司)弹性石英毛细管柱;进样口温度:280 ℃。柱温:初始温度为85 ℃,以15 ℃/min 升温至190 ℃,再以5 ℃/min 升温至225 ℃,然后以20 ℃/min升温至280 ℃,保持5 min;载气:氦气;柱流量:1.0 mL/min,恒流模式;进样模式:不分流;进样体积:1.0 μL。
1.2.2 质谱条件
四级杆温度:150 ℃;离子源温度:280 ℃;传输线温度:280 ℃;电离方式:EI;电离能量:70 eV;扫描时间:0.45 s;监测扫描模式:MRM 模式;碰撞气:氮气,流量为1.5 mL/min;淬灭气:氦气,流量为2.25 mL/min;溶剂延迟:8 min;8种酰胺类除草剂保留时间、定量离子、定性离子和碰撞能量见表1。

表1 8种酰胺类除草剂保留时间、母离子、子离子和碰撞能量
1.3 实验方法
1.3.1 样品采集与运输
用硬质磨口棕色玻璃瓶采集样品。每升水样中加入约100 mg 抗坏血酸,注满样品瓶,上部不留有空间并加盖密封,于4℃以下冷藏、避光保存和运输。采样后在24 h内对样品进行萃取,萃取后若不能及时测定,应于4℃以下避光冷藏,7 d 内完成分析测定。
1.3.2 样品制备
将水样于室温放置,若水样中有细小颗粒物而较为浑浊,可使用0.45 μm水系滤膜过滤,便于固相萃取柱萃取水样品,防止堵塞。
固相萃取过程:依次用5 mL二氯甲烷、5 mL乙酸乙酯、10 mL甲醇、10 mL纯水以3 mL/min流量过柱活化。准确量取500 mL 水样,以15 mL/min 的流量过C18固相萃取柱。用氮气吹干固相萃取柱,去除水分。用5 mL乙酸乙酯以3 mL/min流速洗脱固相萃取小柱,并收集洗脱液。如果洗脱液中含有水分,再用无水硫酸钠去除。将洗脱液置入氮气浓缩仪中,于40 ℃下吹至近干。加入20 μL 内标溶液,用乙酸乙酯复溶定容1.0 mL,上机测定。
1.3.3 样品测定
处理后的样品溶液上机测定,以保留时间定性,色谱峰面积标准曲线内标法定量。
2 结果与讨论
2.1 水样pH值的选择
文献[13]报道,对水样中半挥发性有机物的检测过程中,可通过调节水样的pH来提高化合物稳定性及目标化合物萃取效率。实验考察了以C18为固相萃取柱时pH分别为2、7时的样品萃取效率,结果见图1。结果表明,当pH 7 时8 种酰胺类除草剂的回收率为80.7%~99.2%,均高于pH 2 时的回收率(47.8%~95.2%),因此笔者最终选择调节水样pH 7进行测定。

图1 8种酰胺类除草在不同pH值条件下的回收率(n=6)
2.2 固相萃取柱选择
选择合适的吸附剂对目标化合物进行富集及对干扰物质进行分离至关重要[14]。目前商品化的固相萃取柱种类繁多,主要根据目标化合物理化性质及基体中的主要杂质种类进行固相萃取柱的选择。试验比较了C18、HLB及NH23种固相萃取小柱对样品中目标物的富集和净化效果,对比目标物的回收率,结果如图2 所示。结果表明,NH2柱和HLB 柱只有对双苯酰草胺的回收率大于85%,另7 种酰胺类除草剂样品回收率均低于80%,不能满足实验要求。而经C18柱萃取的样品回收率为85.6%~103.5%,能满足分析要求,因此选择C18柱萃取样品。
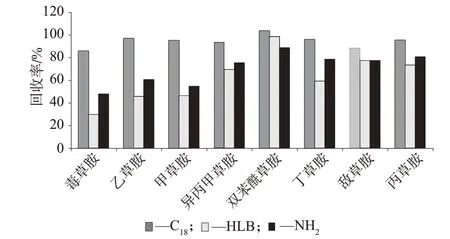
图2 8种酰胺类除草在不同固相萃取条件下的回收率(n=6)
2.3 固相萃取柱活化及洗脱溶剂优化
酰胺类除草剂均为中等极性,实验中8 种目标化合物的极性具有一定差异。样品溶液取得理想的净化效果关键因素取决于合适洗脱溶液和洗脱体积。在洗脱阶段,分别考察了4种溶剂及溶剂组合:5 mL 乙酸乙酯、5 mL 二氯甲烷、5 mL 乙酸乙酯-正己烷(体积比为1∶1)、5 mL 丙酮-正己烷(体积比为1∶1)进行洗脱试验,结果如图3所示。结果表明,使用乙酸乙酯时,C18固相萃取柱吸附的8种酰胺类除草剂回收率均较高(88.6%~102.5%),故选择用5 mL乙酸乙酯洗脱。

图3 8种酰胺类除草在不同洗脱溶液下的回收率(n=6)
2.4 质谱离子源温度的优化
质谱离子源温度是保证化合物离子化效率的关键因素之一。在一定范围内提高离子源温度,可增加化合物的离子化效率,从而提高检测灵敏度,还可以起到净化保护离子源的作用,防止高沸点化合物的残留[15]。分别设定离子源温度为230、250、280 ℃,考察离子源温度对8 种酰胺类除草剂的色谱峰面积的影响,结果见图4。由图4可知,毒草胺、异丙醇草胺、双苯酰草胺3 种除草剂的色谱峰面积随离子源温度的升高显著增大;乙草胺、甲草胺、丁草胺、敌草胺、丙草胺色谱峰面积提高不显著。故选择离子源温度为280℃。
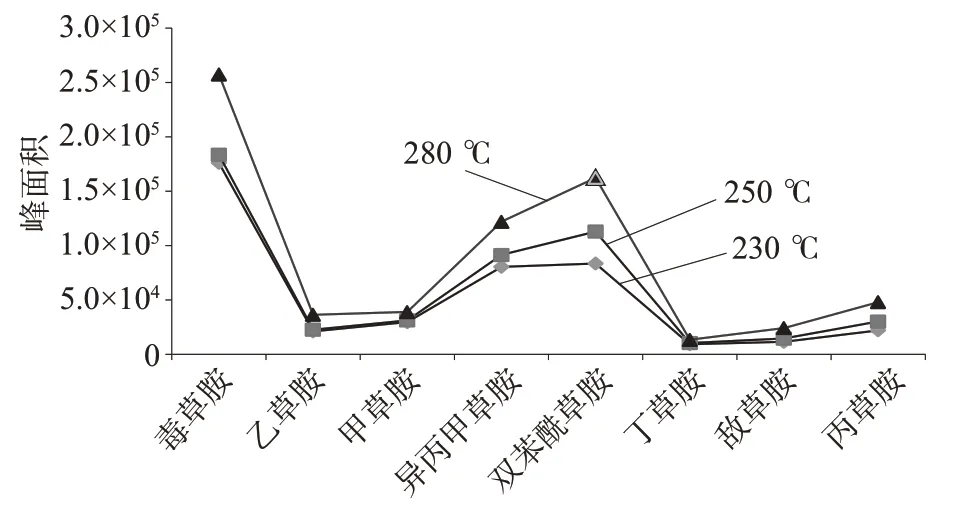
图4 不同离子源温度对应的8种酰胺类除草剂色谱峰面积
2.5 色谱及质谱条件的优化
酰胺类除草剂的分子极性较弱,因此选择极性较小的HP-5MS UI 弹性石英毛细管色谱柱(30 m×0.25 mm,0.25 μm)。优化气相色谱柱温的升温程序,使8 种酰胺类除草剂色谱峰峰形尖锐,对称性好,具有良好的分离度。
先采用全扫描模式在m/z50~550 范围内对8种酰胺类除草剂混合标准溶液进行测定,得到各种酰胺类除草剂的相对保留时间和质谱图。选择分子离子峰或相对丰度较高的特征碎片峰作为母离子。再用离子扫描方式进行二级质谱分析,选择丰度较高的合理断裂碎片离子作为子离子。经优化最终确定8种酰胺类除草剂母离子、子离子和碰撞能量,结果见表1。图5 为8 种酰胺类除草剂标准溶液多反应监测模式总离子流图,图6 为空白样品总离子流图,图7为加标样品总离子流图。

图5 8种酰胺类除草剂标准溶液总离子流图(MRM)
图6 空白样品总离子流图
图7 加标样品加标总离子流图
2.6 线性范围与检出限
精确吸取一定量的8种酰胺类除草剂混合标准溶液,分别加入20 μL内标溶液,逐级用乙酸乙酯稀释,配制成8 种酰胺类除草剂质量浓度均为10~250 μg/L的系列混合标准工作溶液。以8种酰胺类除草剂标准溶液质量浓度与内标物质量浓度的比值为横坐标,除草剂定量离子色谱峰面积和内标物定量离子色谱峰面积的比值为纵坐标,绘制标准曲线,计算线性方程和相关系数。以3倍的信噪比对应的质量浓度作为方法的检出限,10倍的信噪比对应的质量浓度为定量限。
8种除草剂组份线性范围、标准曲线回归方程、相关系数(r)及检出限见表2。

表2 线性关系和方法检出限
2.7 样品加标回收与精密度试验
在空白水样中添加8种酰胺类除草剂混合标准溶液,配制成低、中、高三个浓度水平的加标样品,每种浓度水平配制6 份样品,按1.3 方法进行处理测定,计算加标回收率和相对标准偏差,结果见表3。由表3 可知,8 种酰胺类除草剂回收率为81.1%~104.5%,相对标准偏差为1.6%~6.3%,说明该方法准确度和精密度符合农药残留分析要求。

表3 3种浓度加标回收试验结果(n=6)
3 结语
建立了C18固相萃取柱富集、净化生活饮用水样品中8种酰胺类除草剂,利用气相色谱-三重四级杆串联质谱法检测方法。该方法具有易操作、线性关系良好、准确度和灵敏度高、对环境友好等优点,适用于生活饮用水中多种酰胺除草剂残留检测同时检测。为评价水体污染状况和人体健康风险评估等相关研究提供支持和帮助。
