基于铁死亡和自噬研究酒精的致肝脏细胞损伤作用*
2023-08-07祝子鹤张茜茜张骞骞刘立新徐钧
祝子鹤, 张茜茜, 张骞骞, 刘立新△, 徐钧
基于铁死亡和自噬研究酒精的致肝脏细胞损伤作用*
祝子鹤1, 张茜茜2,3,4, 张骞骞3,4, 刘立新2,3,4△, 徐钧4,5△
(1山西医科大学基础医学院生物化学与分子生物学教研室,山西 太原 030001;2山西医科大学第一医院消化内科,山西 太原 030001;3山西医科大学第一医院科研实验中心,山西 太原 030001;4肝损伤与消化道肿瘤防治委级重点实验室,山西 太原 030001;5山西医科大学第一医院肝胆胰外科,山西 太原 030001)
探索不同浓度酒精作用不同时间对肝细胞、肝星状细胞和肝母细胞瘤细胞铁死亡和自噬的作用及机制。采用不同浓度(0~800 mmol/L)酒精处理AML12小鼠肝细胞株24、48和72 h,处理JS-1小鼠肝星状细胞株和HepG2人肝母细胞瘤细胞株24和48 h;CCK-8法检测细胞活力;油红O染色检测细胞脂质沉积;乳酸脱氢酶(LDH)释放测定细胞损伤水平;Western blot检测细胞活化相关蛋白α-平滑肌肌动蛋白(α-SMA),细胞因子转化生长因子β1(TGF-β1),胶原沉积相关蛋白I型胶原(Col I),铁死亡相关蛋白溶质载体家族7成员11(SLC7A11)、谷胱甘肽过氧化物酶4(GPX4)和铁蛋白重链1(FTH1),以及自噬相关蛋白微管相关蛋白1轻链3(LC3)蛋白水平。(1)酒精抑制AML12细胞活力与时间的延长和浓度的升高相关,脂质沉积呈浓度和时间依赖性增加(<0.05),酒精作用24 h后LDH释放显著增多(<0.01),FTH1和SLC7A11蛋白表达在24 h显著下调(<0.01),LC3-II/LC3-I比值随时间延长而升高(<0.01)。(2)50、100和150 mmol/L酒精处理24 h后JS-1细胞活力增强,而酒精处理48 h后细胞活力则受到显著抑制;50和100 mmol/L酒精处理JS-1细胞24和48 h后TGF-β1、α-SMA和SLC7A11蛋白表达均显著上调(<0.05),各浓度酒精处理后Col I蛋白表达均显著上调(<0.01),GPX4和FTH1表达及LC3-II/LC3-I比值随时间延长而降低(<0.01)。(3)酒精对HepG2细胞活力的抑制作用随时间延长而增强,细胞脂质沉积呈浓度和时间依赖性增加(<0.01),FTH1和GPX4蛋白表达随时间延长而下调(<0.01),随酒精浓度增加而下调(<0.05),酒精处理组SCL7A11蛋白表达和LC3-II/LC3-I比值与对照组相比均显著降低。(1)酒精抑制AML12细胞活力时,铁死亡减弱,自噬增强。(2)低浓度酒精通过增强自噬促进JS-1细胞的活化增殖,而高浓度酒精抑制其活化增殖,且增强SLC7A11下调所致的铁死亡;酒精处理的JS-1细胞自噬强度随时间延长而减弱,铁死亡则依赖时间积累而增强。(3)酒精通过铁死亡增强和自噬受损而抑制HepG2细胞活力和促进脂质沉积。
酒精;肝细胞;肝星状细胞;肝母细胞瘤细胞;铁死亡;自噬
酒精性肝病(alcoholic liver disease, ALD)是指长期持续性酒精摄入引起的肝脏一系列慢性病理改变过程[1]。ALD发病率和死亡率较高,但其发病机制复杂,治疗方法有限,疗效欠佳,因此探索酒精对肝脏细胞损伤的方式和关键生物标志物至关重要[2]。
铁死亡是一种依赖铁和活性氧(reactive oxygen species, ROS)的新型细胞死亡方式,在ALD中发挥重要作用[3]。ALD患者常伴有肝脏铁过载,引起肝脏氧化应激加重、免疫因子释放及肝细胞肿胀[4]。溶质载体家族7成员11(solute carrier family 7 member 11, SLC7A11)、谷胱甘肽过氧化物酶4(glutathione peroxidase 4, GPX4)和铁蛋白重链1(ferritin heavy chain 1, FTH1)是铁死亡发生的关键蛋白,是铁死亡关键的抑制因子[5-6]。过度自噬可导致细胞发生铁死亡[7]。自噬是一种由溶酶体介导的对细胞内异常蛋白质或脂滴等进行降解的过程,微管相关蛋白1轻链3(microtubule-associated protein 1 light chain 3, LC3)是其关键分子。有证据表明自噬在ALD病理过程中起重要作用,但酒精对肝脏自噬激活的影响仍不清楚。
酒精通过破坏机体脂质平衡使肝细胞脂质过度沉积,从而推进氧化应激、炎症反应和纤维化病程[8]。肝星状细胞(hepatic stellate cells, HSCs)活化是肝纤维化的关键。活化的HSCs分泌大量肝纤维化因子,如转化生长因子β1(transforming growth factor-β1, TGF-β1),α-平滑肌肌动蛋白(α-smooth muscle protein, α-SMA)及细胞外基质(extracellular matrix, ECM)成分,如I型胶原(collagen type I, Col I)。人肝母细胞瘤细胞系HepG2维持了一定的肝功能,表型稳定,是替代原代肝细胞研究药物诱导的肝毒性和药物代谢合适的模型[9-10]。所以本研究以AML12(小鼠肝细胞系)、JS-1(小鼠HSCs系)和HepG2细胞为研究对象,探讨不同浓度酒精对多种肝脏细胞在多时间点的铁死亡与自噬的作用。
材料和方法
1 主要试剂
无水乙醇购自国药集团化学试剂有限公司;胎牛血清(fetal bovine serum, FBS)购自Biological Industries;DMEM高糖培养液等细胞培养试剂购自赛文创新(北京)生物科技有限公司;所需抗体购自Abcam。
2 主要方法
2.1细胞培养及分组AML12细胞购自中国科学院上海细胞库,JS-1细胞和HepG2细胞为课题组原有细胞。AML12、JS-1和HepG2细胞分别使用完全DMEM/F12、DMEM低糖和DMEM高糖培养液培养。AML12和HepG2细胞实验组用100和200 mmol/L酒精处理,JS-1细胞实验组用100、150和200 mmol/L酒精处理,处理时间24~72 h。
2.2CCK-8法检测细胞活力将(1~2)×103mL-1的AML12、JS-1和HepG2细胞悬液,以每孔100 μL铺到96孔板中,每组3个复孔,次日换用不同浓度(0~800 mmol/L)酒精处理24~72 h,弃旧培养液,每孔加入含10% CCK-8的完全培养液100 µL,孵育2 h,酶标仪测定450 nm处吸光度,比较不同组间的细胞活力。
2.3油红O染色检测细胞脂质沉积AML12和HepG2细胞接种于24孔板中(每孔0.5×105个细胞),每组6个复孔,按照实验分组处理细胞后,固定液固定25 min,60%异丙醇浸洗5 min,油红O染色液浸染15 min,Mayer苏木素染色液复染1 min。
2.4乳酸脱氢酶(lactate dehydrogenase, LDH)释放测定肝细胞损伤水平同油红O染色处理AML12细胞,取各组上清按照说明书指示进行处理,室温放置5 min,波长450 nm,酶标仪测定吸光度。
2.5Western blot检测蛋白表达提取各组细胞总蛋白,BCA法测定蛋白浓度。100 ℃煮沸使蛋白变性。总蛋白经电泳分离,冰浴转膜,5%脱脂奶粉室温封闭2 h。加入Ⅰ抗4 ℃孵育过夜、Ⅱ抗室温孵育2 h后,增强化学发光法曝光检测相应蛋白的表达信号。用ImageJ软件分析蛋白条带灰度值。
3 统计学分析
采用SPSS和GraphPad Prism 8软件进行统计分析及作图,结果以均数±标准差(mean±SD)表示。组间均数比较采用单因素方差分析,两两比较采用LSD-检验。以<0.05为差异有统计学意义。
结果
1 酒精对AML12、JS-1和HepG2细胞活力的影响
CCK-8实验结果显示,与对照组比较,酒精抑制AML12细胞活力与时间的延长和浓度的升高相关,见图1A; 50、100和150 mmol/L酒精处理24 h后JS-1细胞活力显著增强,而酒精处理48 h细胞活力则随浓度升高而显著受到抑制,见图1B;50和100 mmol/L酒精处理24 h,HepG2细胞活力增强,200、400和800 mmol/L酒精处理24 h,HepG2细胞活力减弱,但酒精处理48 h,HepG2细胞活力比24 h显著减弱,且呈浓度依赖性抑制,见图1C。

Figure 1. Effect of ethyl alcohol (EtOH) on the viability of AML12 (A), JS-1 (B) and HepG2 (C) cells. Mean±SD. n=3. *P<0.05 vs 0 mmol/L.
2 酒精对AML12和HepG2细胞脂质沉积的影响
酒精可使肝细胞产生脂质沉积,油红O染色液能够通过最先浸染脂质来体现出极其微小的脂滴。染色结果显示,与对照组相比,AML12细胞脂质沉积随酒精浓度升高而沉积增多(<0.01),并且随酒精作用时间延长而沉积增多(<0.05),见图2A;酒精处理的HepG2细胞较对照组脂质沉积明显(<0.01),随酒精作用时间延长而沉积增多(<0.01),见图2B。

Figure 2. Effect of ethyl alcohol (EtOH) on the lipid accumulation of AML12 (A) and HepG2 (B) cells (oil red O staining, scale bar=20 μm). Mean±SD. n=6. *P<0.05, **P<0.01 vs control group.
3 酒精对AML12细胞产生LDH的影响
不同浓度酒精干预AML12细胞24~72 h,LDH释放结果显示,与对照组比较,各组细胞LDH释放有不同程度地增加,但酒精作用24 h有显著差异(<0.01),100 mmol/L酒精处理后细胞LDH释放量比200 mmol/L组有增多的趋势,见图3。

Figure 3. Effect of ethyl alcohol (EtOH) on lactate dehydrogenase (LDH) release by AML12 cells. Mean±SD. n=3. *P<0.05, **P<0.01 vs control group.
4 酒精对JS-1细胞中α-SMA、Col I和TGF-β1表达的影响
JS-1细胞以不同浓度的酒精处理24和48 h,Western blot结果显示,α-SMA和TGF-β1在50和100 mmol/L酒精处理下显著升高(<0.05),且随时间延长有下降的趋势,而TGF-β1在150 mmol/L酒精处理下显著下降(<0.05),α-SMA在150 mmol/L酒精处理下有下降的趋势,经各浓度酒精处理不同时间后Col I均显著升高(<0.01),见图4。

Figure 4. Effect of ethyl alcohol (EtOH) on the expression of α-smooth muscle protein (α-SMA), transforming growth factor-β1 (TGF-β1) and collagen type I (Col I) in JS-1 cells. Mean±SD. n=3. *P<0.05, **P<0.01 vs control group.
5 酒精对AML12、JS-1和HepG2细胞铁死亡相关因子SCL7A11、FTH1和GPX4表达的影响
Western blot结果显示,AML12细胞中SCL7A11、FTH1和GPX4蛋白表达在酒精处理48和72 h组均显著上调(<0.01),除GPX4外在酒精处理24 h组显著下调(<0.01),见图5。

Figure 5. Effect of ethyl alcohol (EtOH) on the expression of ferroptpsis-related proteins in AML12 cells. Mean±SD. n=3. *P<0.05, **P<0.01 vs control group.
JS-1细胞中,SLC7A11蛋白表达在50和100 mmol/L酒精处理后显著上调(<0.01),在150 mmol/L酒精处理后显著下调(<0.05),FTH1和GPX4蛋白表达随酒精处理时间的延长而下调(<0.01),见图6。

Figure 6. Effect of ethyl alcohol (EtOH) on the expression of ferroptpsis-related proteins in JS-1 cells. Mean±SD. n=3. *P<0.05, **P<0.01 vs control group.
HepG2细胞中,FTH1和GPX4蛋白表达随酒精处理时间的延长而下调(<0.01),随处理浓度的增加而下调(<0.05);200 mmol/L酒精处理48 h,SCL7A11蛋白表达显著低于100 mmol/L组,亦显著低于24 h(<0.01),见图7。

Figure 7. Effect of ethyl alcohol (EtOH) on the expression of ferroptpsis-related proteins in HepG2 cells. Mean±SD. n=3. *P<0.05, **P<0.01 vs control group.
6 酒精对AML12、JS-1和HepG2细胞自噬相关蛋白LC3表达的影响
Western blot结果显示,与对照组相比,AML12细胞LC3-II/LC3-I比值显著升高(<0.05),且随酒精作用时间延长而升高(<0.01),见图8A;JS-1细胞LC3-II/LC3-I比值除50和100 mmol/L酒精处理48 h有升高趋势,其余组均显著升高(<0.01),且随酒精作用时间延长而降低(<0.01),随酒精作用浓度增加而升高(<0.05),见图8B;与对照组相比,HepG2细胞LC3-II/LC3-I比值均降低,尤其酒精处理48 h显著降低(<0.05),见图8C。
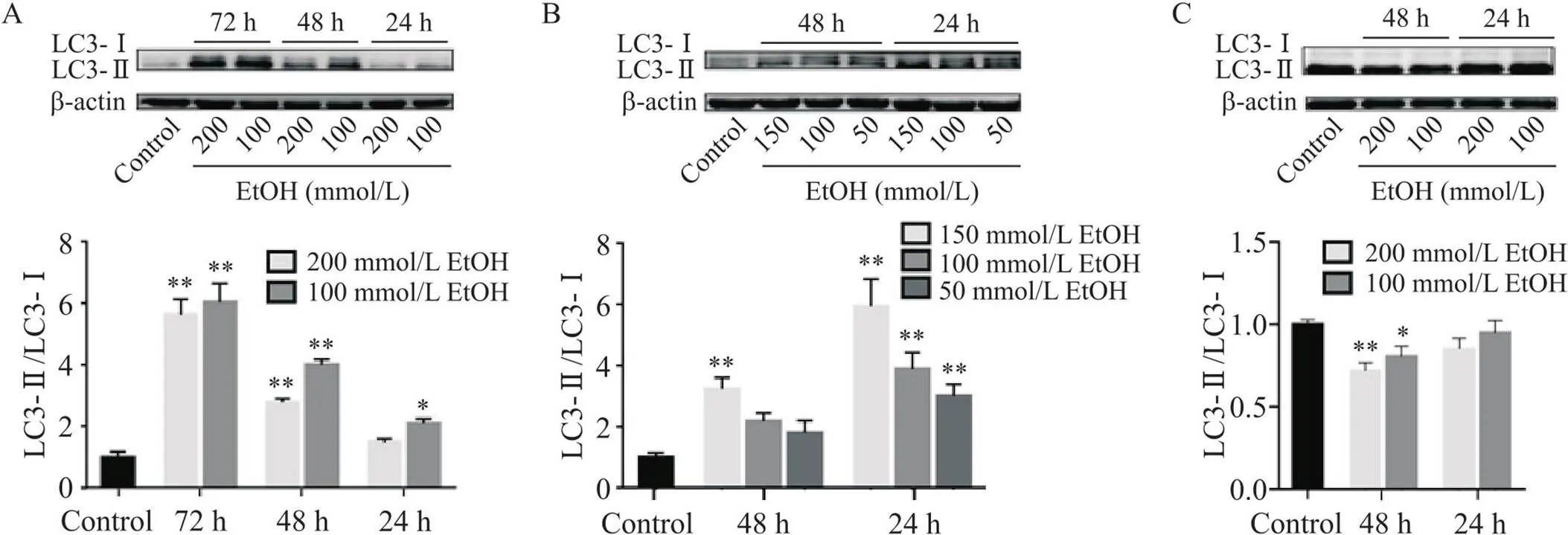
Figure 8. Effect of ethyl alcohol (EtOH) on the expression of autophagy-related proteins in AML12 (A), JS-1 (B) and HepG2 (C) cells. Mean±SD. n=3. *P<0.05, **P<0.01 vs control group.
讨论
酒精主要在肝细胞中代谢,促进甘油三酯合成和外源性脂肪的摄取,刺激肝细胞脂质沉积。以往研究表明,200 mmol/L酒精处理AML12细胞24 h,脂质沉积明显[11];50 mmol/L酒精处理HepG2细胞,亦出现明显的脂质沉积[12]。本研究进一步表明,酒精作用后,AML12和HepG2细胞脂质沉积呈浓度和时间依赖性增多。研究表明,酒精代谢物乙醛(25~200 μmol/L)处理LX2细胞(人HSCs)24 h,细胞被显著激活[13];200 mmol/L酒精处理原代HSCs 48 h,细胞中α-SMA和Col I蛋白表达升高[11]。本研究中,低浓度(50和100 mmol/L)酒精处理JS-1细胞24 h,细胞被活化,释放致纤维化因子,而150 mmol/L酒精处理使细胞活化和释放TGF-β1程度明显减弱。
铁死亡是一种细胞应激,涉及异常的代谢包括铁代谢和脂肪代谢等[14]。据报道,小鼠ALD模型总铁和二价铁明显增多且GPX4和SLC7A11表达下调[15]。本研究显示,AML12经酒精处理24 h后,SLC7A11和FTH1蛋白表达下调,但酒精处理48和72 h后结果逆转。由于细胞死亡是存在多种形式的,所以铁死亡的消减可能有其它细胞死亡方式的添补。也有研究显示,300 mmol/L酒精处理HepG2细胞12 h,GPX4表达水平降低[16]。本研究更加全面地显示铁死亡是随时间延长和浓度升高而增强的,与根据酒精动物模型而预想的结果相一致。据报道,四氯化碳致肝纤维化小鼠模型中,诱导SLC7A11介导的铁死亡可抑制HSCs激活和ECM积累而减轻肝纤维化[17]。在本研究中,GPX4和FTH1蛋白所致铁死亡对JS-1细胞的抑制作用依赖时间延长而增强,而SLC7A11蛋白则仅在150 mmol/L酒精处理下表达下调,与α-SMA和TGF-β1蛋白变化一致。
自噬具有细胞特异性,在肝细胞可调节稳态,但在HSCs则促进其激活[18]。研究表明,在慢性酒精暴露模型中自噬减弱[19],但急性酒精作用下AML12细胞通过ROS生成诱导自噬增强[20]。本研究中AML12细胞自噬随酒精作用时间延长而增加,这可能是肝细胞为缓解急性酒精损伤而产生的细胞应激反应。研究显示,随着酒精浓度的升高(0、50和100 mmol/L),HepG2细胞自噬受损更加严重[21]。本研究进一步表明,HepG2细胞由于酒精所致的自噬受损,不仅随浓度增加,而且也随时间增加。HSCs的激活依赖于自噬降解细胞内的脂滴来提供能量[22],而本研究中JS-1细胞经不同浓度的酒精处理不同时间后自噬水平均升高,但更引人注意的是,自噬强度随酒精作用时间延长而降低。AML12和HepG2细胞均被作为肝脏细胞中的肝细胞进行研究,但二者的结果却相反,HepG2细胞呈现的结果与动物模型相一致,这其中的原因可能是AML12细胞受急性酒精作用更为敏感[20],所以HepG2细胞更合适作为研究铁死亡的酒精细胞模型。总之,本研究结果显示,不同酒精浓度及酒精作用时间对肝脏不同细胞铁死亡和自噬的影响是不同的,为临床上针对不同发展阶段ALD的诊治提供了新思路。但是,具体的相关通路仍需要进一步实验加以阐明。
[1] Buko V, Zavodnik I, Budryn G, et al. Chlorogenic acid protects against advanced alcoholic steatohepatitis in rats via modulation of redox homeostasis, inflammation, and lipogenesis[J]. Nutrients, 2021, 13(11):4155.
[2] Kirpich IA, Warner DR, Feng W, et al. Mechanisms, biomarkers and targets for therapy in alcohol-associated liver injury: from genetics to nutrition: summary of the ISBRA 2018 symposium[J]. Alcohol, 2020, 83:105-114.
[3] Miyata T, Nagy LE. Programmed cell death in alcohol-associated liver disease[J]. Clin Mol Hepatol, 2020, 26(4):618-625.
[4]秦源, 郭永红, 王亚宁, 等. 铁调素调控与肝脏疾病引起的铁代谢紊乱[J]. 肝脏, 2016, 21(6):509-512.
Qin Y, Guo YH, Wang YN, et al. Regulation of hepcidin and iron metabolism disorder induced by liver diseases[J]. Chin Hepatol, 2016, 21(6):509-512.
[5] Tian Y, Lu J, Hao X, et al. FTH1 inhibits ferroptosis through ferritinophagy in the 6-OHDA model of Parkinson's disease[J]. Neurotherapeutics, 2020, 17(4):1796-1812.
[6] Ingold I, Berndt C, Schmitt S, et al. Selenium utilization by GPX4 is required to prevent hydroperoxide-induced ferroptosis[J]. Cell, 2018, 172(3):409-422.e421.
[7] Liu J, Kuang F, Kroemer G, et al. Autophagy-dependent ferroptosis: machinery and regulation[J]. Cell Chem Biol, 2020, 27(4):420-435.
[8] Kisseleva T, Brenner DA. The crosstalk between hepatocytes, hepatic macrophages, and hepatic stellate cells facilitates alcoholic liver disease[J]. Cell Metab, 2019, 30(5):850-852.
[9] López-Terrada D, Cheung SW, Finegold MJ, et al. Hep G2 is a hepatoblastoma-derived cell line[J]. Hum Pathol, 2009, 40(10):1512-1515.
[10] Li X, Xu Z, Bai J, et al. Umbilical cord tissue-derived mesenchymal stem cells induce T lymphocyte apoptosis and cell cycle arrest by expression of indoleamine 2, 3-dioxygenase[J]. Stem Cells Int, 2016, 2016:7495135.
[11] An T, Zhang J, Lv B, et al. Salvianolic acid B plays an anti-obesity role in high fat diet-induced obese mice by regulating the expression of mRNA, circRNA, and lncRNA[J]. PeerJ, 2019, 7:e6506.
[12] Shang Y, Jiang M, Chen N, et al. Inhibition of HMGB1/TLR4 signaling pathway by digitoflavone: a potential therapeutic role in alcohol-associated liver disease[J]. J Agric Food Chem, 2022, 70(9):2968-2983.
[13] Lestari N, Louisa M, Soetikno V, et al. Alpha mangostin inhibits the proliferation and activation of acetaldehyde induced hepatic stellate cells through TGF-β and ERK 1/2 pathways[J]. J Toxicol, 2018, 2018:5360496.
[14] Chen X, Li J, Kang R, et al. Ferroptosis: machinery and regulation[J]. Autophagy, 2021, 17(9):2054-2081.
[15] Zhou Z, Ye TJ, DeCaro E, et al. Intestinal SIRT1 deficiency protects mice from ethanol-induced liver injury by mitigating ferroptosis[J]. Am J Pathol, 2020, 190(1):82-92.
[16] Zhao Y, Lu J, Mao A, et al. Autophagy inhibition plays a protective role in ferroptosis induced by alcohol via the p62-Keap1-Nrf2 pathway[J]. J Agric Food Chem, 2021, 69(33):9671-9683.
[17] Yuan S, Wei C, Liu G, et al. Sorafenib attenuates liver fibrosis by triggering hepatic stellate cell ferroptosis via HIF-1α/SLC7A11 pathway[J]. Cell Prolif, 2022, 55(1):e13158.
[18] Weiskirchen R, Tacke F. Relevance of autophagy in parenchymal and non-parenchymal liver cells for health and disease[J]. Cells, 2019, 8(1):16.
[19] Thomes PG, Trambly CS, Fox HS, et al. Acute and chronic ethanol administration differentially modulate hepatic autophagy and transcription factor EB[J]. Alcohol Clin Exp Res, 2015, 39(12):2354-2363.
[20] Chen C, Wang S, Yu L, et al. H2O2-mediated autophagy during ethanol metabolism[J]. Redox Biol, 2021, 46:102081.
[21] Guo X, Cui R, Zhao J, et al. Corosolic acid protects hepatocytes against ethanol-induced damage by modulating mitogen-activated protein kinases and activating autophagy[J]. Eur J Pharmacol, 2016, 791:578-588.
[22] Thoen LF, Guimarães EL, Dollé L, et al. A role for autophagy during hepatic stellate cell activation[J]. J Hepatol, 2011, 55(6):1353-1360.
Roles of ferroptosis and autophagy in ethanol-induced liver cell injury
ZHU Zihe1, ZHANG Qianqian2,3,4, ZHANG Qianqian3,4, LIU Lixin2,3,4△, XU Jun4,5△
(1,,,030001,;2,,030001,;3,,030001,;4,030001,;5,,030001,)
To investigate the effect of ferroptosis and autophagy on liver cell injury induced by ethyl alcohol (EtOH).Mouse hepatocyte cell line AML12 was treated by various concentrations (0 to 800 mmol/L) of EtOH for 24, 48 and 72 h. Meanwhile, mouse hepatic stellate cell line JS-1 and human hepatoblastoma cell line HepG2 were treated with different concentrations of EtOH for 24 and 48 h. The cell viability was measured by CCK-8 assay. The lipid deposition was detected by oil red O staining. The cells injury level was measured by lactate dehydrogenase (LDH) release. The expression levels of cell activation-related protein α-smooth muscle protein (α-SMA), transforming growth factor-β1 (TGF-β1), collagen deposition-related protein collagen type I (Col I), ferroptosis-related proteins solute carrier family 7 member 11 (SLC7A11), glutathione peroxidase 4 (GPX4) and ferritin heavy chain 1 (FTH1), and autophagy-related protein microtubule-associated protein 1 light chain 3 (LC3) were determined by Western blot.(1) Treatment with EtOH reduced the viability of AML12 cells in a concentration- and time-dependent manner. The lipid deposition was increased in a concentration- and time-dependent manner (<0.05). The LDH level was significantly increased 24 h after EtOH treatment (<0.01). The protein levels of FTH1 and SLC7A11 were significantly decreased 24 h after EtOH treatment (<0.01), while the ratio of LC3-II/LC3-I was increased with increasing EtOH treatment time (<0.01). (2) The viability of JS-1 cells was significantly increased 24 h after treatment with 50, 100 and 150 mmol/L EtOH. In contrast, the activity of JS-1 cells was substantially reduced 48 h after EtOH treatment. The protein levels of TGF-β1, α-SMA, and SLC7A11 were increased (<0.05) in JS-1 cells 24 h and 48 h after treatment with 50 and 100 mmol/L EtOH. Additionally, the protein expression of Col I was significantly increased (<0.01) across all EtOH treatment concentrations. The protein levels of GPX4 and FTH1, and the ratio of LC3-II/LC3-I were decreased with increasing EtOH treatment time (<0.01). (3) The activity of HepG2 cells was significantly reduced after EtOH treatment in a time-dependent manner. Moreover, the lipid deposition was increased in a concentration- and time-dependent manner (<0.01). The protein levels of FTH1 and GPX4 were decreased with increasing EtOH treatment time (<0.01) and concentration (<0.05). The protein level of SLC7A11 and the ratio of LC3-II/LC3-I in EtOH treatment groups were lower than those in control group.(1) Ferroptosis is decreased and autophagy is increased when the viability of AML12 cells is inhibited by EtOH. (2) Low concentration of EtOH promotes the activation and proliferation of JS-1 cells through the enhancement of autophagy, while high concentration of EtOH inhibits the activation and proliferation of JS-1 cells, and promotes the ferroptosis induced by down-regulation of SLC7A11. The autophagy of JS-1 cells is reduced and the ferroptosis is increased after EtOH treatment with increasing time. (3) The viability of HepG2 cells is inhibited and the lipid deposition is promoted through ferroptosis activation and autophagy inhibition.
ethyl alcohol; hepatocytes; hepatic stellate cells; hepatoblastoma cells; ferroptosis; autophagy
1000-4718(2023)07-1265-08
2022-05-18
2023-05-10
刘立新 Tel: 13834238858; E-mail: lixinliu6@hotmail.com;徐钧 Tel: 15103513388; E-mail: xujun@sxmu.edu.cn
R575.1; R363.2
A
10.3969/j.issn.1000-4718.2023.07.013
[基金项目]国家自然科学基金资助项目(No. 81670559);山西省省科技厅自然科学基金项目(No. 202103021224392)
(责任编辑:卢萍,罗森)
