儿童软骨间叶性错构瘤的影像表现及临床病理特征
2023-08-05裴江山汪心韵
裴江山,任 刚,蔡 嵘,汪心韵
(1.泉州市妇幼保健院·儿童医院放射科,福建泉州 362000;2.上海交通大学医学院附属新华医院放射科,上海 200092;3.上海交通大学医学院附属瑞金医院放疗科,上海 200025)
软骨间叶性错构瘤(chondromesenchymal hamartoma,CMH)是一种非常罕见的骨良性间叶性肿瘤,主要治疗方法是外科手术[1-2]。目前关于该病的影像诊断报道较少,且多为个案,临床医生对该病的认识不足,术前容易误诊为恶性肿瘤。笔者收集了8例经手术完整切除后病理证实的患者资料,探讨其影像学表现特点,旨在提高对该病的认识及诊断,避免临床误诊与不当治疗。
1 资料与方法
1.1 一般资料
收集上海交通大学医学院附属新华医院2012年12月至2021年12月经手术病理证实的8例CMH患者的临床资料,其中4例病变位于鼻部,另4例位于胸壁肋骨。
1.2 方法
1.2.1影像学检查
患儿检查前口服水合氯醛0.5 mL/kg进行镇静睡眠。8例患儿中,3例同时行CT和磁共振成像(MRI)检查,3例仅行CT检查,2例仅行MRI检查。CT检查采用西门子SOMATOM Definition扫描仪,扫描层厚3~5 mm,增强扫描碘对比浓度300~350 mgI/mL,注射总量2 mL/kg,注射速率1.0~1.5 mL/s,采集动脉期及实质期。MRI检查采用Siemens Prisma 3.0T MRI扫描仪。平扫序列包括常规T1WI、T2WI加脂肪抑制;增强扫描横断面、矢状面及冠状面T1WI加脂肪抑制序列,对比剂采用钆喷酸葡胺(Gd-DTPA),注射剂量为0.1 mmol/kg。
1.2.2病理学检查
所有病例均行完整手术切除病灶,标本经10%中性甲醛固定,石蜡包埋,常规切片,苏木精-伊红(HE)染色,制片后光镜下观察。免疫组织化学检查采用EnVision两步法,标记抗体包括波形蛋白(Vimentin)、S-100、平滑肌肌动蛋白(SMA)、细胞膜表面黏糖蛋白(EMA),1例同时行DICER1、USP6、NTRK基因检测。
1.3 临床数据整理与图像分析
分析8例CMH患者的性别、年龄、发病部位、临床表现、术前诊断、免疫组织化学检查结果等临床病理特征,以及病变的位置、分布、与周围组织关系、是否有钙化或骨化、是否有出血囊腔和强化特点等影像学特征。图像由2名高年资放射诊断医师独立阅片分析,意见不一致时共同讨论直至达成一致意见。增强CT病变强化程度的评估:CT值增加10~30 HU记为轻度;增加>30~50 HU为中等;增加>50 HU记为明显强化。
2 结 果
2.1 临床特征
8例患儿中男6例,女2例,男女比例为3∶1。发病年龄从出生1 d至3岁,平均年龄为9.4个月,诊断时最小年龄为17 d(出生时即发现)。4例病变位于鼻部,临床表现包括鼻腔、鼻窦肿物伴呼吸困难、睡眠打鼾、眼球突出等;另4例位于胸壁肋骨,临床上均表现为胸壁局部肿块,术前1例误诊为骨纤维结构不良。
2.2 病理特征
2.2.1形态
鼻部病变肿块呈灰白色,质脆,局部可见软骨组织。肋骨病变最大径1.5~6.0 cm,切面呈灰红色,质软,局部囊性伴出血,部分区域伴钙化。
2.2.2镜下观察
肿瘤主要为透明软骨,部分伴骨化,间质纤维母细胞增生,核分裂象少见。肋骨内病变血管扩张充血,局部见破骨样多核巨细胞,可见类似动脉瘤样骨囊肿的出血性囊腔。
2.2.3免疫组织化学检查结果
免疫组织化学检查结果显示软骨细胞Vimentin(+)7例、S-100(+)7例、梭形间质细胞SMA(+)4例、EMA(+)2例,见表1。1例行DICER1、USP6、NTRK基因检测,结果均为阴性。

表1 CMH的临床病理特征
2.3 CT、MRI表现
4例鼻部病变中1例行CT检查,1例行MRI检查,2例同时行CT和MRI检查,典型病例见图1~3。其中3例CT均为单发病灶,表现为鼻腔或筛窦区域混杂密度肿块,3个病灶内均可见钙化,局部向眼眶内侵袭,其中1例同时累及眼眶、鼻咽部及左侧上颌窦(图1A、1B);1例CT增强后肿块呈中等强化(图2A、2B)。3例MRI表现为T1WI低、T2WI高信号为主的混杂信号软组织肿块,病灶T2WI内可见条状低信号提示钙化(图3A),增强后病变均呈明显不均匀强化(图1D、2D、3C、3D),1例肿块向前颅底延伸(图2D)。
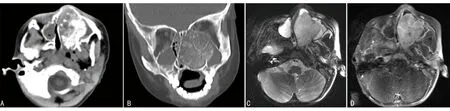
A:CT轴位显示左侧鼻腔混杂密度肿块,局部累及左侧上颌窦及鼻咽部;B:冠状位骨窗显示病变局部向左眼眶内突出,内可见轮辐状钙化;C:T2WI显示肿块呈混杂高信号,其内条状低信号提示钙化;D:T1WI增强后病变明显强化。
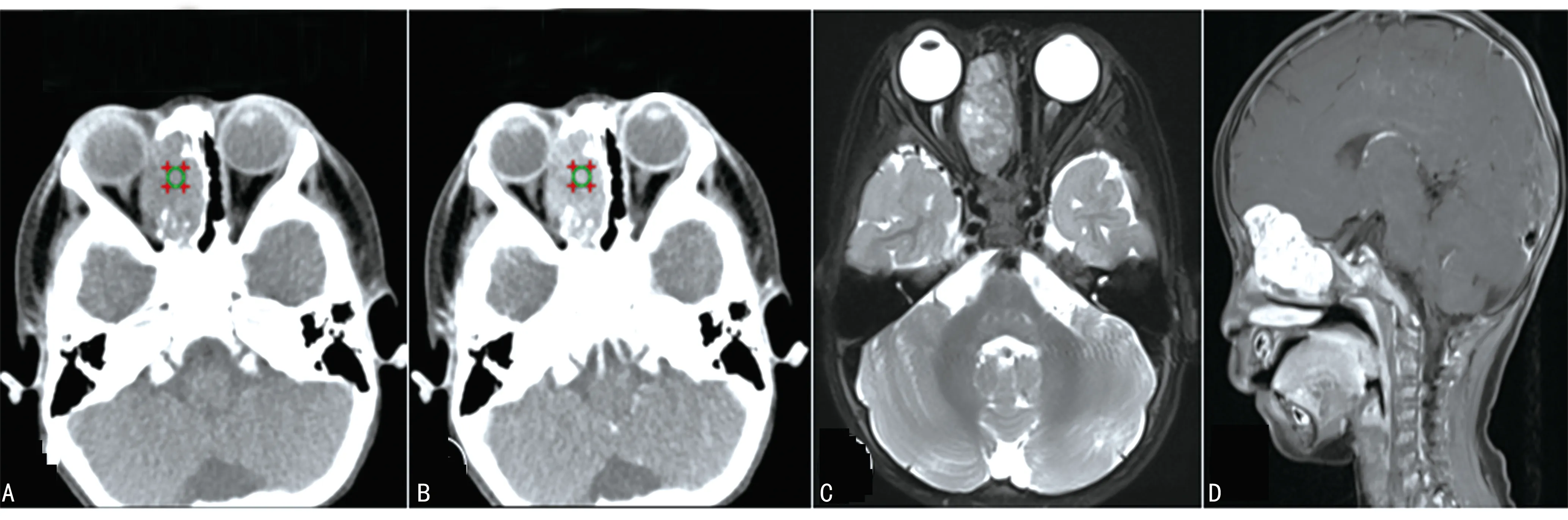
A:CT平扫显示右侧鼻腔混杂密度肿块,CT值约44 HU,其内可见砂粒状钙化,病变局部向右侧眼眶内突出;B:CT增强后呈中等强化,CT值约为75 HU;C:T2WI显示肿块呈混杂信号;D:T1WI增强后呈明显强化,肿块局部向前颅底延伸。
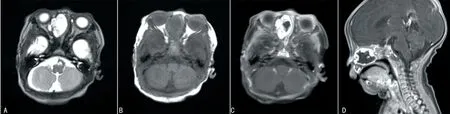
A:轴位T2WI显示右侧鼻腔及筛窦混杂信号肿块,其内条状低信号提示钙化可能,肿块局部向右眼眶内侧壁突出,鼻中隔左偏;B:T1WI显示肿块呈等低信号;C、D:T1WI增强后病变周边呈明显强化,中央部分无强化。
4例胸壁病变均来源于肋骨,2例行CT检查,1例行MRI检查,1例同时行CT和MRI检查,典型病例见图4、5。其中2例单发,2例多发。CT表现为边界清楚的膨胀性骨质破坏,伴边缘为主的钙化或骨化(图4A、5A),局部可突向胸腔压迫肺组织,虚拟重建技术(VR)显示肋骨膨胀呈筛网状改变(图4C),增强后1例呈中等到明显强化,2例肿瘤实性部分强化不明显(图4B);2例MRI检查T2WI显示肿块呈囊实性混杂信号,1例可见典型的出血性液-液平面(图5C),呈继发性动脉瘤样骨囊肿改变。结果见表2。

A:CT平扫显示左侧第8、9肋骨膨胀性骨质破坏伴混杂密度软组织肿块,其内可见多发边缘型钙化,以肋骨面为著;B:CT增强后显示肿瘤实性部分强化不明显;C:VR显示左侧肋骨膨胀及筛网状改变。
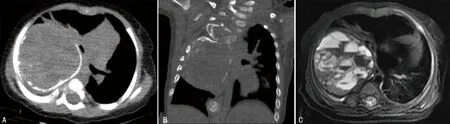
A:CT平扫显示右侧胸部混杂密度肿块,边缘可见典型蛋壳样钙化,病变向右侧胸腔及纵隔内生长;B:CT冠状位显示右侧多根肋骨呈膨胀性骨质破坏改变,并与肿块相连;C:T2WI显示肿块呈囊实性混杂信号,其内可见多发出血囊腔及典型的液-液平面。
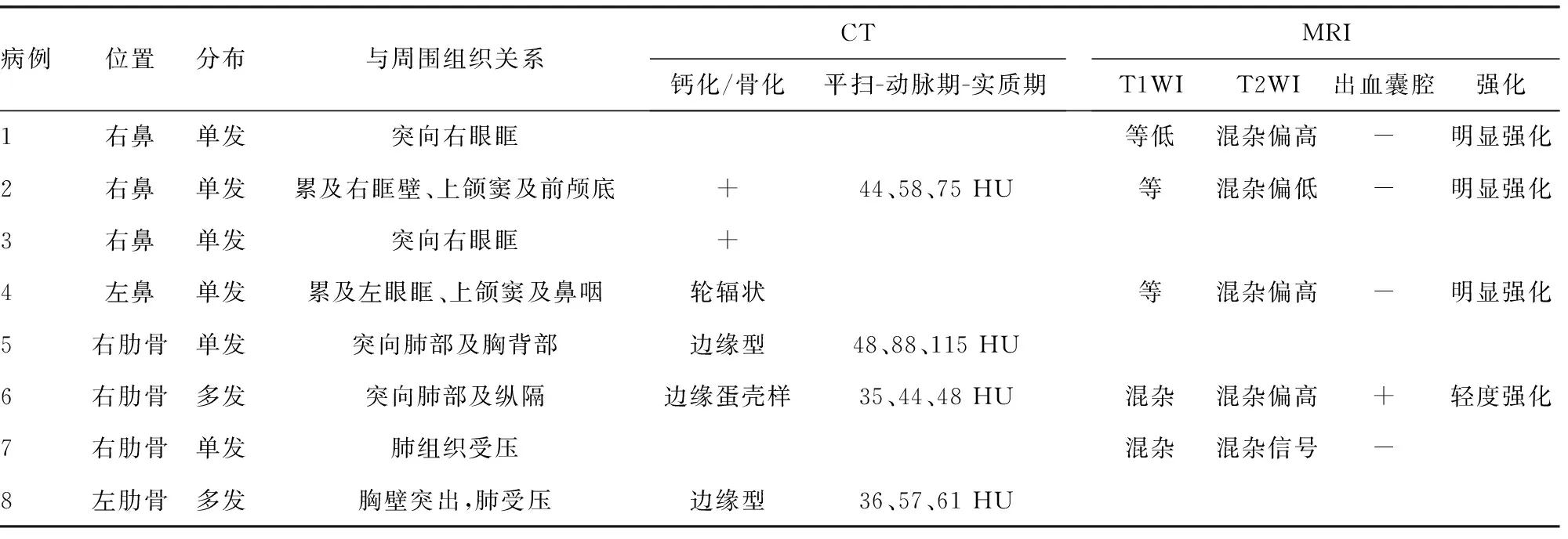
表2 CMH的CT、MRI表现
3 讨 论
1979年,MCLEOD和DAHLIN提出了间叶性错构瘤这一术语[3],CMH较多发生于新生儿或婴幼儿,是一种非常罕见的骨良性间叶性肿瘤[4],以往称为“软骨性错构瘤”“间叶瘤”和“婴儿骨软骨瘤”等,2013版WHO骨肿瘤与软组织分类将NCMH与MHCW一起归入未明确肿瘤性质的肿瘤这一类病变中,并统一使用名称CMH[5]。2016年WHO头颈部肿瘤分类第4版[6]首次将其收录其中,2020版WHO骨肿瘤分类[7]将MHCW归为骨的其他间叶性肿瘤。
CMH的典型发病部位包括鼻和胸壁肋骨[8-9],据文献统计,NCMH约占1/3,MHCW约占2/3。本研究病例有4例位于鼻部筛窦,均为单发且突向眼眶生长,其中2例累及上颌窦,1例累及前颅底,1例累及鼻咽;另4例起源于胸壁肋骨,其中2例为单发,2例多发,均压迫邻近肺组织,1例累及纵隔。CMH最常见于婴幼儿或新生儿,部分发生于胸壁的病例在产检时被发现[4,10],也有发生于成人的罕见病例。本研究病例中NCMH和MHCW各4例,男6例,女2例,男女比例为3∶1。发病年龄从出生1 d至3岁,平均年龄为9.4个月。4例NCMH临床症状为鼻腔、鼻窦肿物伴呼吸困难、睡眠打鼾,4例MHCW均表现为胸壁局部突出的肿块。
CT和MRI是CMH的首选检查技术,两者相结合有助于确定肿物的位置、特征、解剖关系及侵蚀情况。NCMH的CT表现为囊实性混杂密度软组织肿块影,伴或不伴有钙化,增强后病变呈明显不均匀强化[11]。钙化可能是NCMH与其他鼻部肿瘤鉴别的重要依据。一项针对NCMH病变CT影像特征的回顾性研究[12]显示67%的NCMH可见骨质变薄或侵蚀,53%显示筛窦或通过筛板向颅内延伸,50%内部可见钙化,40%可见囊性成分,67%有中到明显强化。MRI显示T1WI呈混杂低信号,T2WI呈混杂高信号,增强后呈不均匀显著强化。本研究中4例NCMH,3例CT检查见明确钙化,1例MRI检查T2WI内可见条片状低信号,亦提示钙化可能;4例均可见向眼眶内侵蚀,1例累及右眶壁、上颌窦及前颅底,1例同时累及左眼眶、上颌窦及鼻咽。由于肿块的明显强化及对临近骨质的侵袭破坏,术前1例误诊为血管瘤,1例误诊为鼻腔的恶性肿瘤。
MHCW典型的CT表现为起源于肋骨的囊状膨胀性骨质破坏,边界清楚,几乎所有病变可见钙化或骨化,部分可见出血性囊腔及液-液平面(继发性动脉瘤样骨囊肿)[13];MRI所见征象与CT几乎相同,但在显示动脉瘤样骨囊肿方面优于CT[14]。本研究4例MHCW均来源于肋骨,3例CT检查表现为边界清楚的膨胀性骨质破坏伴边缘为主钙化,1例MRI可见继发性动脉瘤样骨囊肿改变。有文献报道[4,15]免疫组织化学染色虽可提示病变内各成分来源,但没有特异性的标记物,对诊断的意义较小。本组病例免疫表型结果显示软骨细胞主要表达Vimentin和S-100蛋白,梭形间质细胞表达SMA。
本病在影像学检查中部分呈现破坏性的生长方式,术前容易被误诊为其他恶性肿瘤。根据发病部位和累及范围,NCMH主要应与鼻筛脑膨出、横纹肌肉瘤及软骨肉瘤等相鉴别[11]。鼻筛脑膨出通常表现为没有明显强化的混合软组织密度肿块,也有骨缺损,但没有破坏前颅窝[16]。放射学上很难将NCMH与横纹肌肉瘤和软骨肉瘤区分开来,但后两者往往生长迅速且边界不清,更具破坏性[17]。MHCW的鉴别诊断包括原发性动脉瘤样骨囊肿、骨软骨瘤、软骨肉瘤和内生性软骨瘤等[18]。原发性动脉瘤样骨囊肿表现为膨胀性、分叶状骨质破坏,MRI可见囊性区伴周围低信号环[19],组织学上缺乏实性的软骨结节;骨软骨瘤好发于长管状骨干骺端、背向关节生长,髓腔与母骨相通,与MHCW明显不同;软骨肉瘤主要发生于成人和老人软骨内化骨的骨骼,以股骨和胫骨最为多见,婴幼儿罕见;内生性软骨瘤儿童相对少见,常表现为溶骨性骨质破坏,可见骨内扇贝征,基质骨化较罕见[20]。
本研究的局限性在于纳入的样本量较少,且仅1例做了DICER1基因检测,针对近些年文献报道的CMH与致病胚系DICER1基因突变及其肿瘤家族(包括胸膜肺母细胞瘤、卵巢性索间质瘤、囊性肾瘤和松果体母细胞瘤等)的相关性[21-22],还需扩大样本量及完善DICER1基因检测进一步研究分析。此外,本组鼻软骨间叶错构瘤仅1例做CT增强检查,CT强化特征未总结。
综上所述,CMH多见于婴幼儿鼻部与胸壁肋骨,影像学主要表现为混杂密度或信号的软组织肿块,常伴钙化或骨化,部分可见继发性动脉瘤样骨囊肿的出血性囊腔,具有一定特征性。该病具有良性肿瘤的生物学行为,术前的准确诊断对患儿的治疗和预后至关重要。
