基于二代和三代转录组测序揭示甘蔗重要亲本对黑穗病菌侵染的响应机制
2023-07-20罗正英李纯佳吴转娣李旭娟刘新龙
胡 鑫 罗正英 李纯佳 吴转娣 李旭娟 刘新龙
基于二代和三代转录组测序揭示甘蔗重要亲本对黑穗病菌侵染的响应机制
胡 鑫 罗正英 李纯佳 吴转娣 李旭娟 刘新龙*
云南省农业科学院甘蔗研究所/ 云南甘蔗遗传改良重点试验室 / 农业农村部甘蔗生物学与遗传育种重点实验室(云南), 云南开远 661699
黑穗病是甘蔗生产中的主要病害, 严重影响甘蔗产量。解析甘蔗重要亲本与黑穗病菌相互作用的分子机制及筛选抗病基因对抗黑穗病优良品种的培育具有重要意义。本研究选用我国甘蔗育种中的重要亲本新台糖ROC25 (抗黑穗病)及其姊妹系ROC22 (感黑穗病)为研究对象, 采用单分子实时测序技术(三代测序)和二代转录组测序技术分析和鉴定2个亲本感染黑穗病菌后的转录组谱及差异转录本。三代转录组测序分析共获得79,885条转录本序列, 其中含60,115条完整开放阅读框、3692个可变剪接事件、1799个LncRNA、29,139个SSR和7794个转录因子, 共有74,066个转录本得到注释, 占总数的92.72%。通过对二代测序数据分析, 在抗病亲本中筛选出9716个差异转录本, 在感病亲本中筛选出2033个差异转录本。差异转录本的GO和KEGG富集分析结果表明抗病亲本中共富集到的GO条目和KEGG通路均要多于感病亲本, 且植物MAPK信号通路、苯丙素生物合成、植物-病原互作、亚油酸代谢和蔗糖和淀粉代谢等代谢通路在抗感亲本中均被显著富集, 为抗感亲本对黑穗病的共同抗病途径。进一步, 对植物MAPK信号通路的分析表明, MAPK超家族基因成员在抗感亲本中呈现不同的表达方式, 在抗病亲本中具有更多差异表达的和的转录本, 且、和基因仅在抗病品种中发生显著表达变化, 推测其可能与亲本的抗病表型关联。此外, 抗感亲本中众多WRKY、MYB、NAC和AP2/ERF-ERF等抗病相关转录因子响应了黑穗病菌胁迫, 且主要表现为上调表达。与感病亲本相比, 抗病亲本显示出更多的差异表达转录因子, 推测这些抗病亲本特有的转录因子可能对防御黑穗病菌具有积极作用。本研究完善了新台糖亲本基因组注释信息, 为解析新台糖优异亲本与黑穗病菌互作机制以及抗黑穗病基因资源的挖掘利用提供指导。
甘蔗; 黑穗病菌; 全长转录组; RNA-seq; 新台糖
甘蔗(spp. hybrids)是全球最重要的糖料作物, 也是重要的新型可再生能源作物。黑穗病是甘蔗生长的主要病害, 在世界各大蔗糖生产国均有发生, 为害极为严重, 造成甘蔗产量的减少, 被称为甘蔗的“癌症”[1]。甘蔗黑穗病的致病菌为黑穗病菌(), 其病原孢子(smut teliospore)可通过土壤、空气、雨水和昆虫等传播途径侵染蔗芽或侧枝, 刺激顶端分生组织快速生长, 至后期茎顶端组织四周被大量黑穗病孢子包裹并形成典型的黑色鞭状物。新生成黑鞭中的病菌孢子会再次侵染快速生长的顶芽和侧芽, 从而加剧病情发生和传播[2]。研究表明, 黑穗病的严重程度既取决于甘蔗品种抗性, 又与环境条件密切相关, 易感品种在种植2~3年内表现出非常高的发病率(>60%的发病率), 而高抗品种在试验期间表现出接近零的发病率[3-4]。提高甘蔗品种的抗性是预防和控制黑穗病最为经济有效的方式[5-6]。然而, 由于甘蔗杂交重组率极低、抗黑穗病遗传基础复杂, 导致传统杂交育种培育抗黑穗病甘蔗品种难度较大。如果利用优异甘蔗种质, 深入研究甘蔗与黑穗病菌互作机制, 尽可能多地筛选和鉴定控制甘蔗抗黑穗病的关键基因, 有望将其应用于甘蔗抗黑穗病的分子育种, 加快抗黑穗病品种的选育进程。在甘蔗与黑穗病菌互作中, 致病孢子主要通过侵染蔗芽进入芽顶端分生组织定植, 该过程6~36 h即可完成[7]。对感黑穗病甘蔗品种而言, 在病原侵染后3~4周后, 即可在茎顶端分生组织观察到大量黑穗病菌丝, 而在抗病品种顶端分生组织中检测不到或仅有极少量菌丝, 表明抗感品种中黑穗病发病进程存在明显差异[8]。感病品种约在感染120 d后开始形成黑鞭, 黑鞭四周含有大量厚垣孢子, 而在茎顶端下部节间薄壁细胞中分布有黑穗病菌丝, 不形成孢子[9]。目前, 世界各甘蔗生产国大都采用甘蔗株发病率来评价甘蔗抗黑穗病的抗性等级, 田间评价调查周期通常持续7~8个月[10]。早期, Dean等[11]首先把甘蔗抵御黑穗病机制划分为侵染抗性(芽抗性)和定植抗性(内部组织抗性)。后来, 普遍将甘蔗抗黑穗病机制分为外部抗性和内部抗性等两类, 外部抗性包括甘蔗芽结构特征、芽体鳞片的厚度或紧密性、芽体内苯丙酸和糖基类黄酮等化学物质造成的物理化学屏障, 阻止黑穗病菌进入芽内部建立感染; 而内部抗性是甘蔗响应黑穗病菌侵染后采取的主动抵御致病菌生长的一序列防御反应[12-15]。随着基因组学、转录组学和蛋白组学等技术应用于甘蔗与黑穗病菌互作研究, 甘蔗抗黑穗病机制逐渐被揭示。众多研究表明甘蔗接种黑穗病菌后, 细胞壁强化途径(如木质素合成)、次级代谢产物合成(如糖蛋白、植保素、多胺、类黄酮等)、植物与病原菌的相互作用、活性氧、丝裂原活化蛋白激酶(Mitogen-activated Protein Kinase, MAPK)信号通路、植物激素信号通路以及与抗性相关的其他调控途径的基因表达发生明显变化, 且抗病品种和感病品种中基因表达模式也出现明显的差异, 表明它们与甘蔗响应黑穗病菌侵染密切相关, 在调控甘蔗抵御黑穗病反应中发挥重要作用[16-21]。甘蔗与黑穗病菌互作进程伴随着广泛的生理和分子(基因、蛋白、小RNA和降解组、代谢组等)水平变化, 抗性水平受到多个主效基因、众多微效基因的共同控制[10,22]。最新研究发现, 转录因子[23]、Ras-类似GTP结合蛋白基因[24]、茉莉酸生物合成的关键基因[25]和抗氧化酶系[26]对甘蔗抗黑穗病起正调控作用, 而转录因子对甘蔗响应黑穗病菌胁迫具负调控作用[27]。这些关键抗黑穗病基因的鉴定增进了人们对甘蔗抗病机制的理解, 有望推进甘蔗抗黑穗病分子育种的进程。但是, 迄今为止, 人们对甘蔗如何精准防御黑穗病侵染的分子机制的了解还很有限, 有待进一步深入。
新台糖甘蔗系列品种是19世纪80年代从我国台湾地区引进的优良品种, 由于高产、高糖和适应性好等特性, 它们迅速发展成为中国大陆地区的主栽品种, 高峰期种植面积超过90%, 成为我国最为重要的主栽品种, 强有力地支撑了中国大陆地区蔗糖产业的快速发展[28]。同时, 由于其具有良好的育种特性, 新台糖甘蔗系列品种也成为我国最为重要的甘蔗亲本, 其后代如柳城05-136、柳城03-182、桂糖42、桂糖49、云蔗08-1609、云蔗05-51已成为我国主栽的当家甘蔗品种。在我国甘蔗黑穗病愈发严重的背景下, 新台糖亲本与黑穗病菌互作机制具有重要的研究价值。鉴于此, 本研究选用新台糖系列亲本中最为知名的2个亲本(ROC22和ROC25), 开展其响应黑穗病侵染的转录组学研究。2个优良亲本属于姊妹系, 都是台糖69-463的子代[29-30], 遗传背景较为相似[31], 但在抗黑穗病性上表现出明显的差异, ROC22易感黑穗病[32], 而ROC25高抗黑穗病[33]。我们希望采用遗传背景相似但抗性差异显著的亲本更有利于揭示新台糖亲本响应黑穗病侵染的分子遗传学基础, 筛选可供开发利用的抗病基因, 为后续更好地利用新台糖亲本培育高产高糖抗黑穗病品种提供重要的指导。
1 材料与方法
1.1 试验材料
所用甘蔗亲本材料ROC22和ROC25均由国家甘蔗种质资源圃(云南开远)提供。人工接种鉴定试验所用黑穗病菌采自云南省农业科学院甘蔗研究所科研第一基地、第二基地和第三基地种植的ROC22亲本。采集的黑色鞭状物于室温自然风干4~5 d后收集病鞭上冬孢子, 充分混匀后分装于小滤纸袋中, 保存于4℃冰箱中备用。接种前先检查取冬孢子活力,取微量混合冬孢子经1%水琼脂培养(含50μg mL–1氨苄青霉素) 12 h后镜检, 萌发率大于90%的冬孢子即为合格的接种菌。
1.2 黑穗病菌侵染
取ROC25和ROC22成熟期种茎, 切成单芽段后在室温下流水冲洗24 h。单芽段在室温下干燥后转移到光照培养箱(12 h光照-12 h黑暗循环, 32℃, 80%湿度)中催芽。待萌动芽长度约2 cm时准备接种黑穗病菌。选用合格的冬孢子配置成浓度为5×106孢子mL–1的黑穗病孢子悬浮液, 用无菌注射器吸取菌液注射入芽基部, 并以清水注射为对照组。处理后的种茎重新移到光照培养箱中继续培养。根据苏亚春[34]报道, 在人工针刺法接种黑穗病菌12 h、24 h、48 h和168 h的4个时间段, 在48 h以内的3个时段抗感材料蔗芽内部的黑穗病菌拷贝数量未有明显差异, 但到168 h时达到十分显著差异, 表明抗感材料响应黑穗病菌侵染的最活跃时期应在48 h以后。鉴于此, 本研究选择黑穗病菌接种后5 d (120 h)、8 d (192 h)和11 d (264 h)作为取样时间点, 以芽顶端分生组织为取样部位。每一个样品由3个芽顶端分生组织混合组成, 并设置3个生物学重复, 加上清水处理共计36个样品(表1)。样品取完后立刻用液氮速冻后保存与–80℃冰箱。使用干冰运输至百迈克生物科技有限公司(中国青岛)开展三代和二代转录组测序。
1.3 转录组测序和质量评估
由百迈克生物科技有限公司(中国青岛)完成文库构建和转录组测序。二代转录组36个样品, 使用Illumina平台进行建库和测序。三代全长转录组使用36个样品的混样来建库, 共使用1个cell, 使用PacBio仪器进行全长转录组测序[35]。通过全长序列识别、isoform水平聚类得到一致序列和一致序列校正(polishing)最终得到非冗余转录本序列[36-37]。考虑被侵染组织样品中含有黑穗病菌, 将转录组数据比对黑穗病菌基因组, 去除比对上的转录本[21], 利用BUSCO对最终获得的转录组进行完整性评估[38]。
1.4 全长转录本结构分析
参考Liu等[39]采用BLAST version 2.2.26软件对所有序列进行两两比对来鉴定可变剪接事件。筛选500 bp以上的转录本, 利用MISA (MIcroSAtellite identification tool, http://pgrc.ipk-gatersleben.de/misa/)软件做SSR分析。使用TransDecoder软件(https:// github.com/TransDecoder/TransDecoder/releases)从转录本序列中识别可靠的潜在编码区序列(Coding Sequence, CDS)。应用最广泛的编码潜能分析方法对转录本进行lncRNA的预测, 包括Coding Potential Calculator (CPC)分析[40]、Coding-Non-Coding Index (CNCI)分析、pfam蛋白结构域分析和Coding Potential Assessment Tool (CPAT)分析[41]等。使用iTAK软件对植物转录因子预测、分类并统计分析[42]。使用BLAST软件将得到的非冗余转录本序列与NR、Swissprot、GO、COG、KOG、Pfam、KEGG和eggNOG数据库比对, 获得转录本的注释信息。
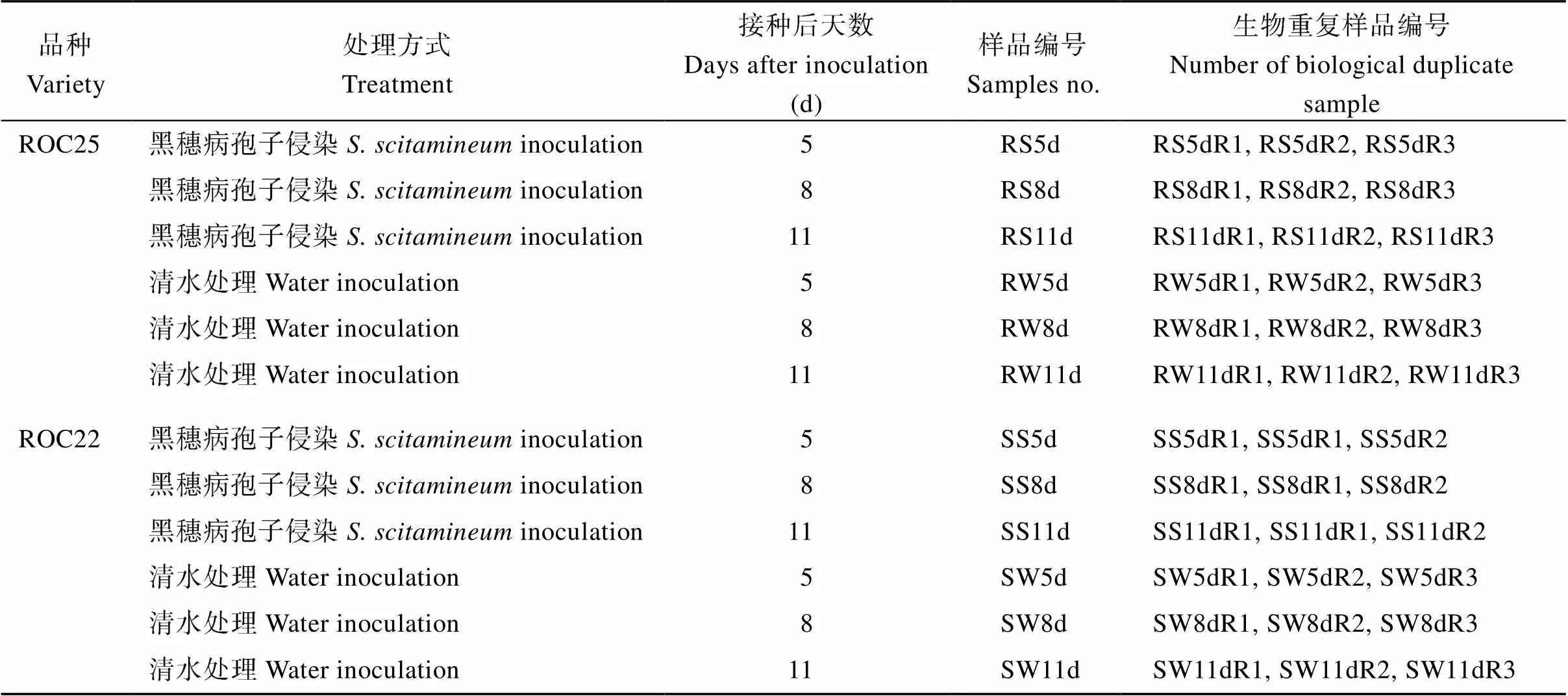
表1 黑穗病菌接种设计和转录组测序样品采取
1.5 差异转录本筛选、GO富集分析和KEGG富集分析
使用三代全长转录本为参考, 利用STAR[43]将二代转录组获得Clean Reads与全长转录本进行序列比对, 获取转录本的位置信息。使用RSEM软件[44]对转录本的表达水平进行定量。将黑穗病侵染样品与同一时间点的清水处理样品构成一个差异基因分析组, 共形成6个对比组, 即感病亲本ROC22获得3个比较组(SS5d-vs-SW5d、SS8d-vs-SW8d、SS11d- vs-SW11d)和抗病亲本ROC25获得3个比较组(RS5d-vs-RW5d、RS8d-vs-RW8d、RS11d-vs-RW11d)。依据各对比组的FPKM值, 将Fold Change≥2且FDR < 0.05作为筛选差异基因的标准, 使用DEseq2[45]软件进行差异基因分析。
1.6 实时荧光定量PCR验证
取抗感亲本黑穗病菌侵染和清水处理5 d、8 d和11 d的蔗芽生长点部位组织, 通过液氮将样品在研钵中充分研磨成粉后, 每份样品取约100 mg粉末于1.5 mL离心管中进行总RNA的提取。将提取的总RNA合成第1链cDNA并进行实时荧光定量PCR反应, 反应程序为95℃ 3 min; 95℃ 10 s, 57℃ 30 s, 40个循环。从4个常用的内参基因、、和[46]中筛选表达相对较为稳定的基因作为最终内参基因。随机选择15个转录本, 根据转录组序列设计特异性引物, 所有基因引物序列信息表2。使用各时间点黑穗病侵染组与清水对照组转录本表达量倍数FC (FPKM)和实时荧光定量PCR表达量差异倍数FC (2–DDCt)开展皮尔逊相关系数分析, 检测转录本数据和荧光定量PCR数据的相关性。
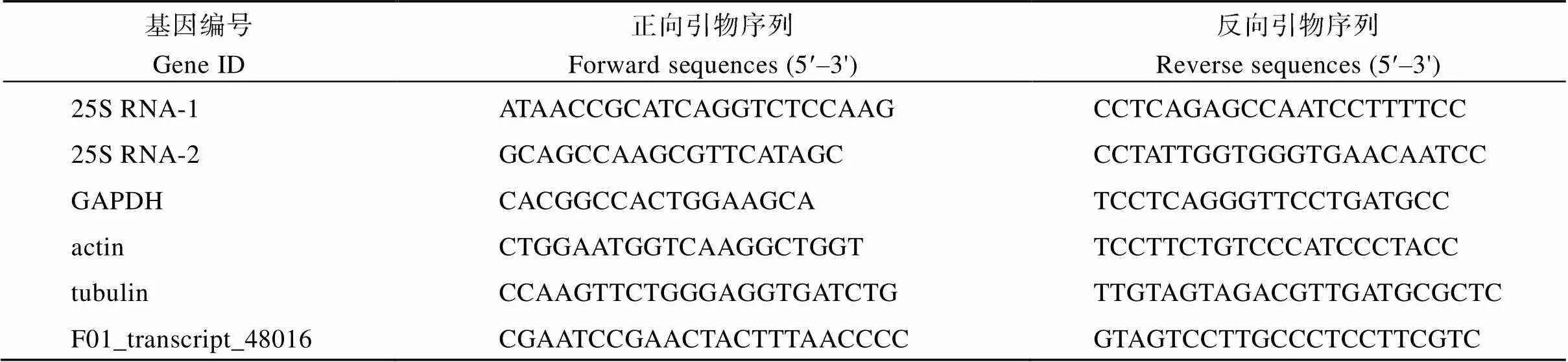
表2 实时荧光定量PCR的引物序列
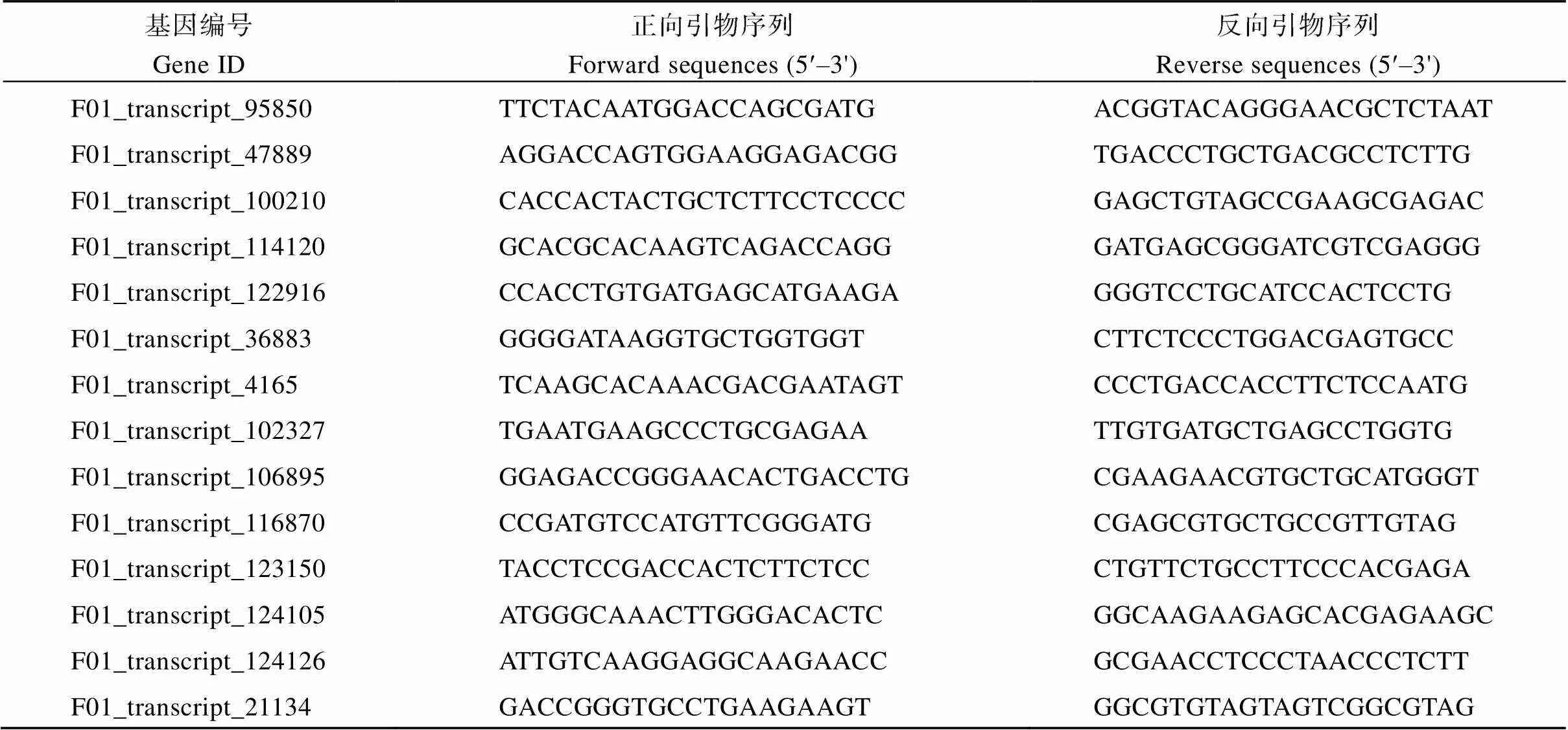
(续表2)
2 结果与分析
2.1 转录组测序数据统计
使用PacBio平台对抗黑穗病亲本ROC25和感黑穗病亲本ROC22在病菌侵染和清水的不同时段共计36个样品的混样进行全长转录组测序, 共获得22.13 Gbp的clean data。使用Illumina seq对36个样品开展二代转录组测序, 共获得211.53 Gbp的clean data, 各样本的Q30均超过95%, 整体质量较高。对全长转录组获得的clean data进行环形一致性序列分析, 获得288,335个CCS序列, 其中包含全长非嵌合(FLNC, full length readsnon-chimeric)序列262,407条, 对FLNC序列一致性聚类和Polish分析获得高质量一致性序列123,473条, 使用CD-HIT软件去除冗余序列后得到82,801条转录本序列。除比对上黑穗病菌基因组的转录本, 最终获得79,885条转录本, 转录本序列平均长度为1990 bp (图1-A)。利用BUSCO对转录本进行完整性评估(图1-B), 单拷贝、多拷贝及不完全覆盖的基因占比达82.92%, 表明所获得的全长转录本具有较高的完整度。
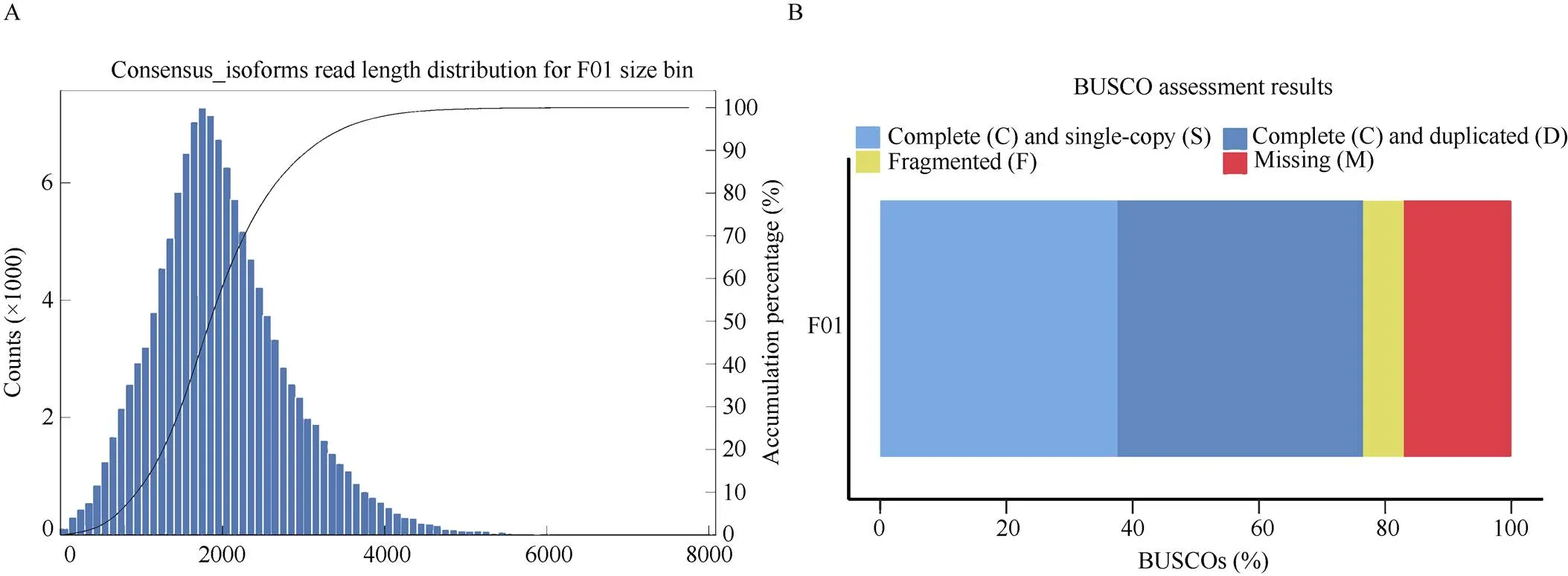
图1 全长转录组测序一致性序列度长分布和完整度评估
A为全长转录组测序获得的一致性序列的长度分布, 左侧Y坐标轴对应一致性序列长度频数分布直方图, 右侧Y坐标轴对应一致性序列长度累积频率曲线。B为全长转录组测序获得的一致性序列的完整性评估结果。
A: the read length of consensus isoform, the left Y-axis corresponds to the frequency distribution histogram of consensus isoform read length, and the right Y-axis corresponds to the cumulative frequency curve of consensus isoform read length. B: the assessment of sequence annotation completeness.
2.2 全长转录本可变剪接、编码区、SSR位点、LncRNA和转录因子预测
本研究共鉴定到3692个可变剪接事件, 分布在2871个基因中, 其中发生一次AS事件占总比78% (图2-A); 鉴定到了1799个LncRNA (图2-B), 其中529个LncRNA预测到靶基因; 共鉴定到29,139个SSR, SSR类型大部分都是单核苷酸(P1)、2个核苷酸(P2)或3个核苷酸(P3)的重复(图2-C)。编码区预测共获得80,363个ORF, 其中完整ORF (即同时预测到起始密码子和终止密码子)有60,115条, 完整ORF编码蛋白序列长度范围为0~2500个氨基酸, 平均氨基酸长度为341个, 分布最多的范围为100~200个(图2- D)。在所有转录本中共预测到8002个转录因子, 分属219个家族, 其中预测转录因子数量最多的是RLK-Pelle_DLSV家族(406个), 其次为bHLH家族(269个)、C2H2家族(193)、MYB-related家族(193)、C3H家族(180个)、GRAS家族(175个)和bZIP家族(174个) (图2-E), 表明这些转录因子积极参与调节新台糖亲本生长及抗病反应过程。
2.3 全长转录本功能注释分析
对得到的非冗余转录本序列在NR、Swissprot、GO、COG、KOG、Pfam、KEGG和eggNOG等八大主要基因或蛋白数据库进行比对和注释发现, 共有74,066个转录本得到注释(表3), 占总数的92.72%, 其中在Nr数据库得到注释的转录本最多, 达73,781个, 占比达92.36%, 其次为eggNOG数据库(70,639个, 占比达88.43%)和GO数据库(62,274个, 占比达77.95%)。
从Nr注释来看(图3-A), 有55.51%的转录本注释到甘蔗的近缘属种高粱, 26.63%的转录本注释到玉米, 仅有3.63%的转录本注释到甘蔗杂交种, 源于甘蔗基因组数据极不丰富。从eggNOG注释情况来看(表4), 50.81%的基因归入功能未知基因, 其次为信号传导机制和翻译后修饰、蛋白质转换, 分子伴侣相关基因, 占比分别为6.97%和6.87%。从GO注释情况来看(图3-C), 在细胞组分上注释最多的GO条目是细胞和细胞器及其膜部分, 在生物过程注释最多的GO条目是代谢过程、细胞进程和单组织等过程, 在分子功能方面注释最多的GO条目是催化活性和蛋白绑定, 表明这些GO功能在全长转录组占据主导。从KEGG注释来看(图3-B), 代谢途径主要集中在全局和总览图、碳水化合物代谢、、脂质代谢和氨基酸代谢通路, 遗传信息过程主要集中在翻译和折叠、分类和降解通路, 环境信号过程主要集中在信号传导途径通路, 细胞过程主要集中在运输和分解代谢通路, 有机系统主要集中在环境适应通路。

图2 甘蔗全长转录本特征和结构分析
A为基因的可变剪接事件发生次数分析; B为CPC、CNCI、pfam蛋白结构域和CPAT等4种分析方法预测的LncRNA的韦恩分析; C为SSR类型分布作图, c为混合微卫星(2个SSR距离小于100 bp), p1为单碱基, p2为双碱基, p3为三碱基, p4为四碱基, p5为五碱基, p6为六碱基; D为预测的CDS编码蛋白长度分布图; E为排名前20的转录因子类型。
A: the classification of AS cases; B: the overlap of long non-coding RNAs predicted by four different method; C: the classification of SSR types, c, p1, p2, p3, p4, p5, and p6 represent compound SSR, mono-nucleotide, di-nucleotide, tri-nucleotide, tetra-nucleotide, penta- nucleotide, and hexa-nucleotide, respectively. D: the length distribution of predicted protein; E: the top 20 types of predicted transcription factors.
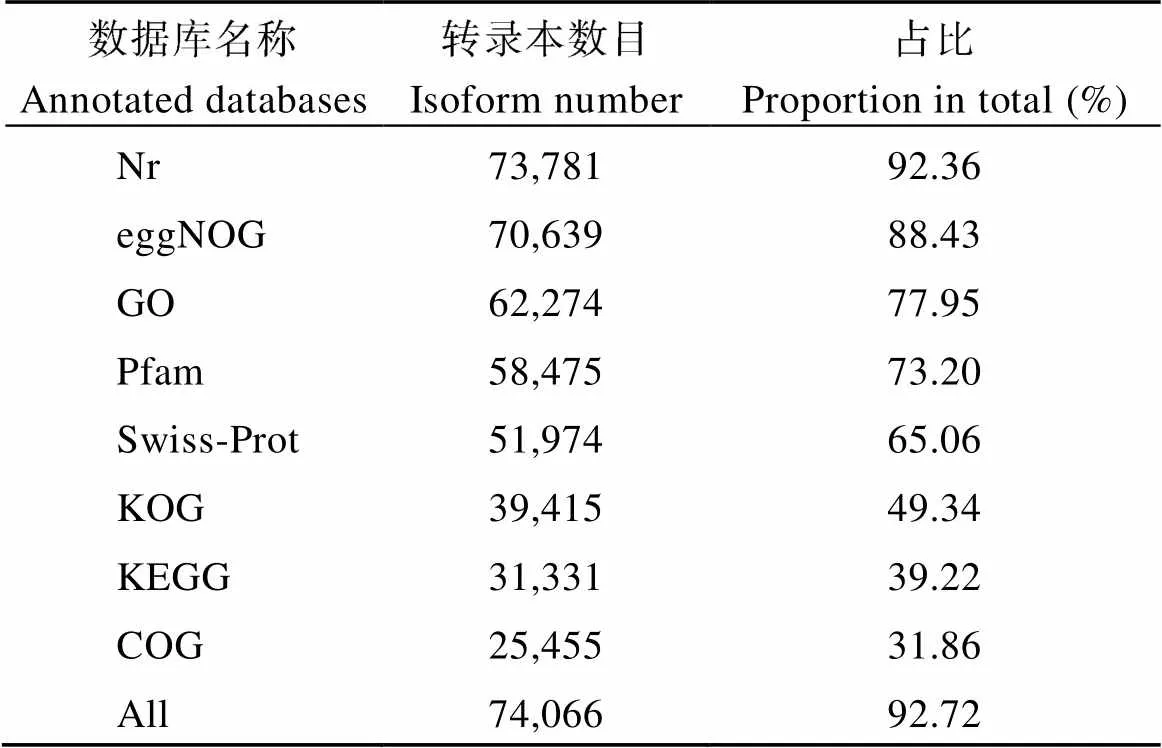
表3 全长转录本在不同数据库中注释数量统计

图3 甘蔗全长转录本注释及分类统计
A、B和C为Nr、KEGG和GO注释分类统计图。
A, B, and C indicate Nr, KEGG, and GO characteristics of the obtained transcripts, respectively.

表4 全长转录本eggNOG注释分类统计

A、B、C和D分别为ROC22清水处理生物学样品、ROC22黑穗病侵染生物学样品、ROC25清水处理生物学样品和ROC25黑穗病侵染生物学样品的相关性系数; E和F分别为18个ROC22样品和18个ROC25样品的PCA分析图。
A, B, C, and D indicate the correlation coefficient of test samples from ROC22 inoculated with water, ROC22 inoculated with, ROC25 inoculated with water, and ROC25 inoculated with, respectively. E and F indicate PCA diagram of ROC22 and ROC25 samples, respectively.
2.4 差异基因筛选
对所有36个二代转录组样品开展皮尔逊相关系数分析发现, 各基因型在接种黑穗病菌或清水条件下的生物学重复之间的相关性系数都大于0.90 (图4-A~D), 表明各条件组内生物学重复样品之间相关程度很高, 重复结果可靠。对各基因型样品的二代测序结果进行主成分分析(Principal Component Analysis, PCA)发现, 抗感亲本同一时间点清水处理和黑穗病侵染样品表达量在PCA图上呈现不同的空间分布, 表明二者表达量存在差异, 适合开展差异基因分析。
相比较清水处理, 感黑穗病亲本ROC22接种黑穗病菌后5 d、8 d和11 d时分别筛选到1829、139和101时筛选到1829个差异表达基因, 数量上呈现逐步降低的趋势; 抗黑穗病亲本ROC25侵染5 d、8 d和11 d时分别筛选到927、4240和5824个差异表达基因, 数量上呈现逐渐增加的趋势。整体来看, 侵染5 d时, 感病亲本相比较抗性亲本获得更多的差异表达基因, 对侵染的响应表现的更为活跃, 但随着侵染时间的延续, 感病亲本响应活跃度出现大幅下降, 而抗病亲本响应活跃度呈现显著增强的趋势, 表现出有更多的基因参与响应; 从上下调差异表达基因数来看, 无论是抗病还是感病亲本, 都呈现出侵染后上调基因多于下调基因, 尤其侵染的第一第二阶段, 表明上调表达基因在侵染的早中期扮演着十分重要的角色。抗感亲本黑穗病侵染后获得的总差异表达基因进行维恩图分析(图5-B~D)发现, 抗感亲本共有1010个差异表达基因, 其中810个上调表达, 30个下调表达, 170个上/下调趋势不同; 感病亲本特有1023个差异表达基因, 包括994个上调表达基因和199个下调表达基因; 抗病亲本特有8706个差异表达基因, 其中5359个上调表达, 3517个下调表达。
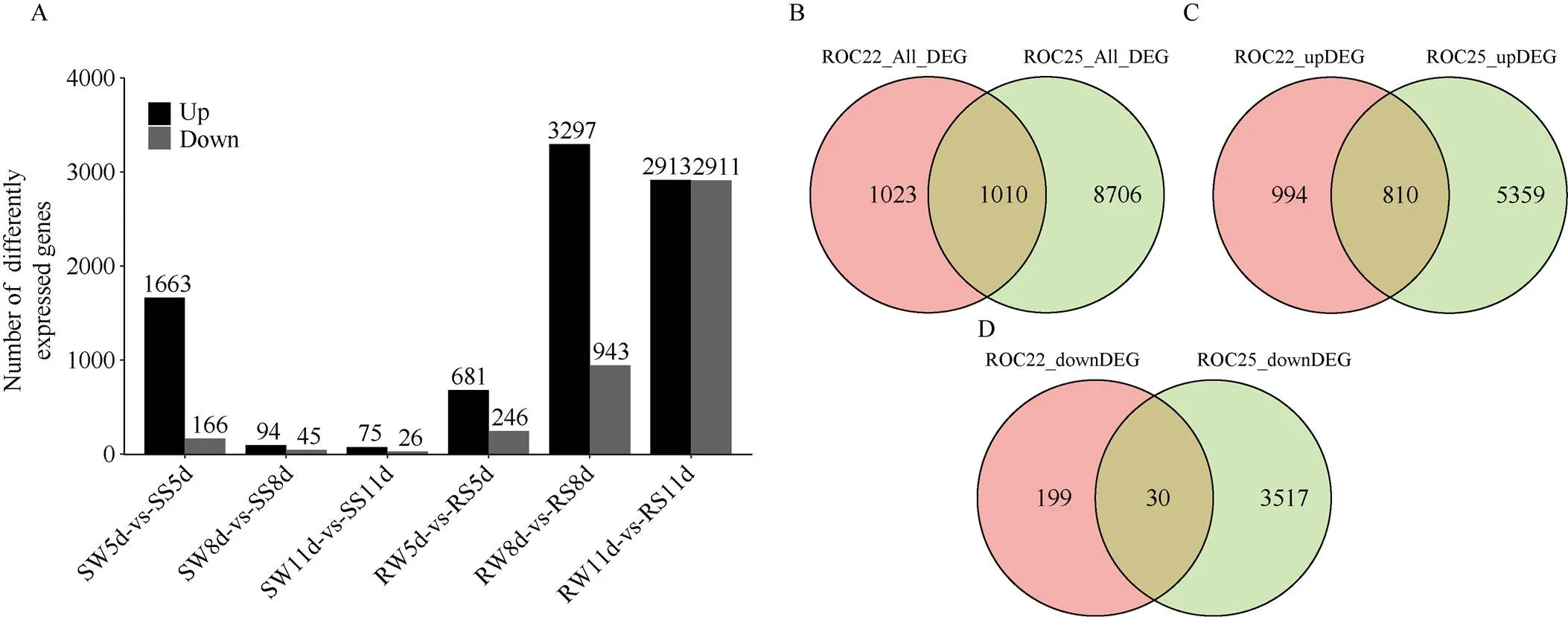
图5 ROC22和ROC25不同侵染阶段差异基因数量统计和两亲本间差异基因韦恩分析
A为ROC22和ROC25感染黑穗病后5 d、8 d和11 d差异基因数量; B为不同侵染阶段2个亲本差异基因维恩图。
A: the differentially expressed transcripts (DETs) in ROC22 and ROC25 at different days afterinfection; B: the overlap of DEGs between ROC22 and ROC25.
2.5 实时荧光定量PCR验证转录组测序结果
随机选取15个转录本通过实时荧光定量PCR对基因表达水平进行定量分析, 并将各时间点每个接种黑穗病菌的甘蔗亲本与同时间点清水对照组的表达量倍数(FC(2-DDCt)和FC(FPKM))进行相关性分析。15个基因实时荧光定量PCR和转录组表达量变化情况如图6所示, 定量结果与转录组测序结果大体趋势相似, 但部分组间的基因表达趋势存在一定差异, 可能原因是检验的基因表达水平较低或者基因序列特征复杂。将RNA-seq和qPCR的表达水平差异倍数取对数后进行线性回归分析(附图1), 结果显示两者相关系数为0.6941 (<0.05), 表明测序结果与qPCR结果相关性较好, 测序结果较为可靠。
2.6 差异表达基因GO分类和富集分析
感病亲本中2033个差异表达基因被显著富集到205个GO条目, 包括105个细胞过程条目、90个分子功能条目和10个细胞组分条目; 富集程度排名前20的条目中细胞过程富集因子最大的是防御反应(GO:0006952)、蛋白磷酸化(GO:0006468)和羟脂蛋白代谢过程(GO:0031407), 分子功能富集因子最大的是蛋白激酶活性(GO:0004672)、蛋白丝氨酸/苏氨酸激酶活性(GO:0004674)和磷酸转移酶活性(GO:0016773); 细胞组分富集因子最大的是膜(GO:0016020)、细胞外周(GO:0071944)和膜内在成分(GO:0031224) (图7-A)。
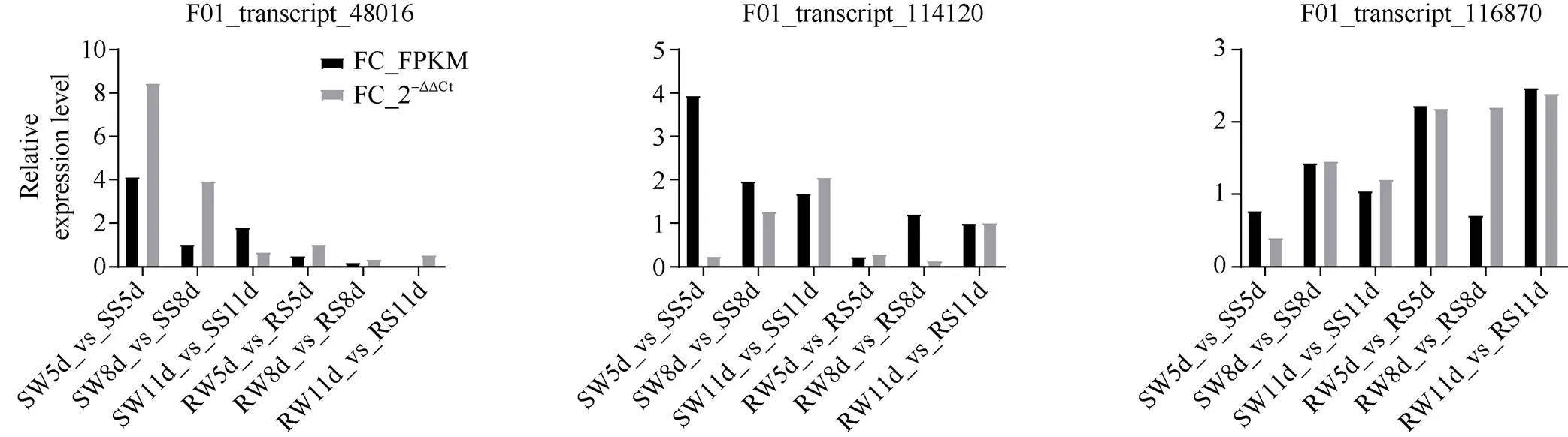
(图6)
对15个转录本(表2)比较各个接种病菌组与清水对照组的的RNA-seq的FPKM值和实时荧光定量PCR中2–ΔΔCt值的差异倍数。
15 transcripts were used in this validation referring to Table 2. Fold changes of each RNA-seq expression data (FPKM value) and quantitative real-time PCR (2–ΔΔCt) betweeninoculation group and water inoculation group were compared.
抗病亲本9716个差异表达基因共富集到1083个GO条目, 其中细胞过程878个条目, 细胞组分73个条目, 分子功能203个条目; 富集程度排名前20的条目中细胞过程富集因子最大的是核小体装配(GO:0006334)、染色质组装(GO:0031497)和DNA包装(GO:0006323); 分子功能富集因子最大的是蛋白质异二聚活性(GO:0046982)、四吡咯结合(GO:0046906)和转录因子活性(GO:0003700)等; 细胞组分富集因子最大的是核小体(GO:0000788)、DNA包装复合物(GO:0044815)和蛋白质-DNA复合物(GO:0032993)等(图7-B)。
对获得的富集GO条目开展维恩图分析(图7-C~ E), 在生物过程, 抗病亲本有151个特有富集条目, 感病亲本有38个特有富集条目, 抗感亲本共有差异表达基因共有52个特有富集条目; 在细胞组分中, 抗病亲本有63个特有富集条目, 感病亲本无特有富集条目, 抗感亲本共有差异表达基因共有10个特有富集条目; 在分子功能中, 抗病亲本有534个特有富集条目, 感病亲本有37个特有富集条目, 抗感亲本共有差异表达基因共有68个特有富集条目。
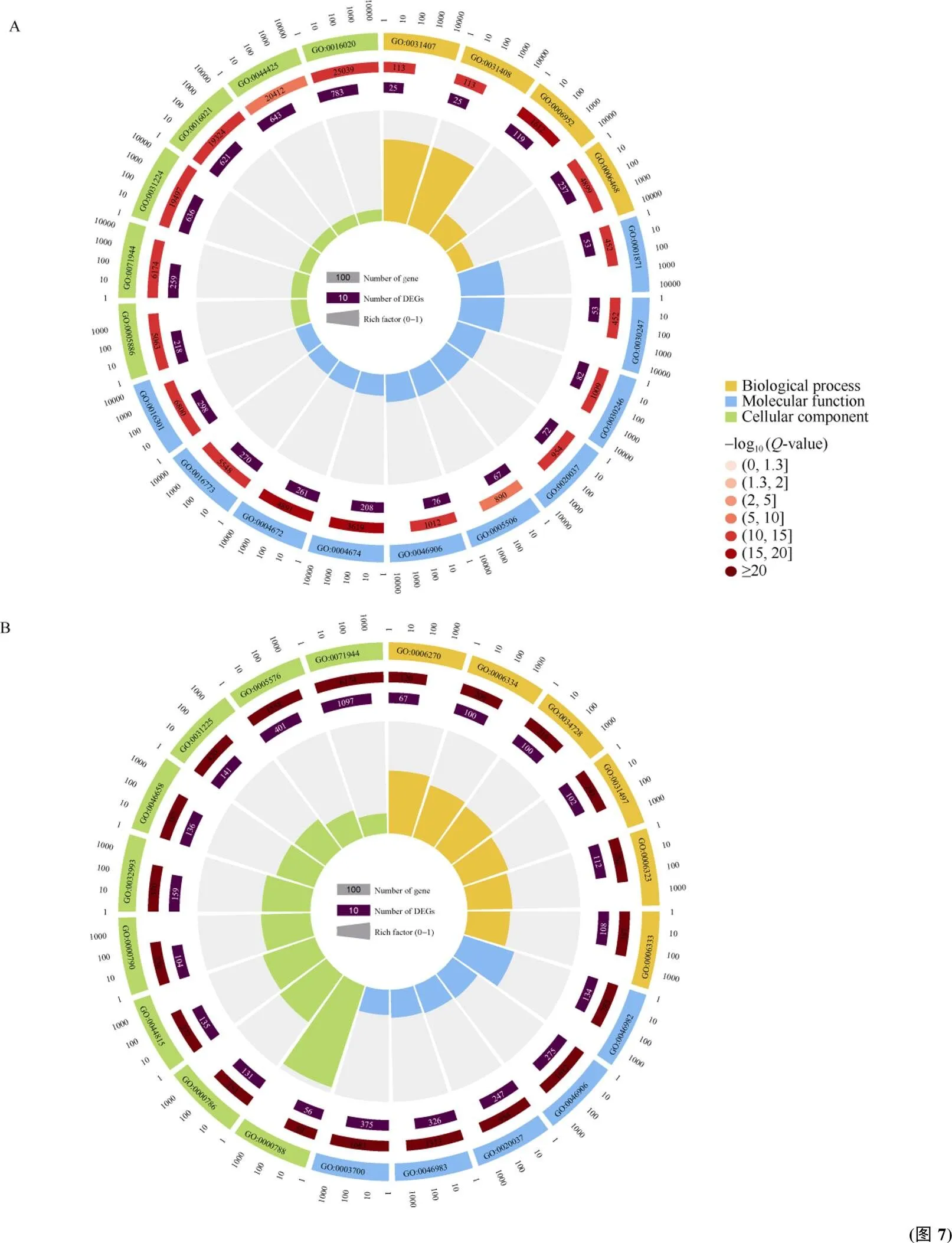
A和B分别为 ROC22差异基因和ROC25差异基因富集程度排名前20的GO条目。C、D和E为抗感亲本中差异基因富集的生物过程(MP)、细胞组分(CC)和分子功能(MF)条目比较分析。
A and B indicate TOP20 GO enrichment terms of differentially expressed transcripts in ROC22 and ROC25, respectively. C, D, and E indicate comparison of biological processes (MP), cellular components (CC), and molecular functions (MF) in different varieties, respectively.
2.7 差异表达基因KEGG分类和富集分析
感病亲本中2033个差异表达基因被显著富集到17个KEGG通路, 其中次级代谢物生物合成(Biosynthesis of secondary metabolites)、亚油酸代谢(Linoleic acid metabolism)、苯丙素生物合成(Phenylpropanoid biosynthesis)、植物-病原互作(Plant-pathogen interaction)和植物MAPK信号通路(MAPK signaling pathway-plant)的显著性水平最高(图8-A)。抗病亲本9716个特有差异表达基因经被显著富集到27个KEGG通路, 其中植物MAPK信号通路、植物激素信号转导(Plant hormone signal transduction)、苯丙素生物合成(Phenylpropanoid biosynthesis)、植物-病原互作和亚油酸代谢的显著性水平最高(图8-B)。
将抗感亲本中差异基因富集的KEGG通路进行维恩图分析(图8-C)发现, 12个通路属于两者共有, 包括植物MAPK信号通路、苯丙素生物合成、植物-病原互作、亚油酸代谢、淀粉和蔗糖代谢等。感病亲本特有5个KEGG通路, 分别为丙烷、哌啶和吡啶类生物碱的生物合成(Tropane, piperidine and pyridine alkaloid biosynthesis)、黄酮类生物合成(Flavonoid biosynthesis)、玉米素生物合成(Zeatin biosynthesis)、叶酸生物合成(Folate biosynthesis)和囊泡运输中SNARE相互作用(SNARE interactions in vesicular transport)。抗病亲本特有15个KEGG通路, 它们主要是植物激素信号转导、光合作用、DNA复制(DNA replication)、类胡萝卜素生物合成(Carotenoid biosynthesis)和光合作用生物碳固定(Carbon fixation in photosynthetic organisms)等。
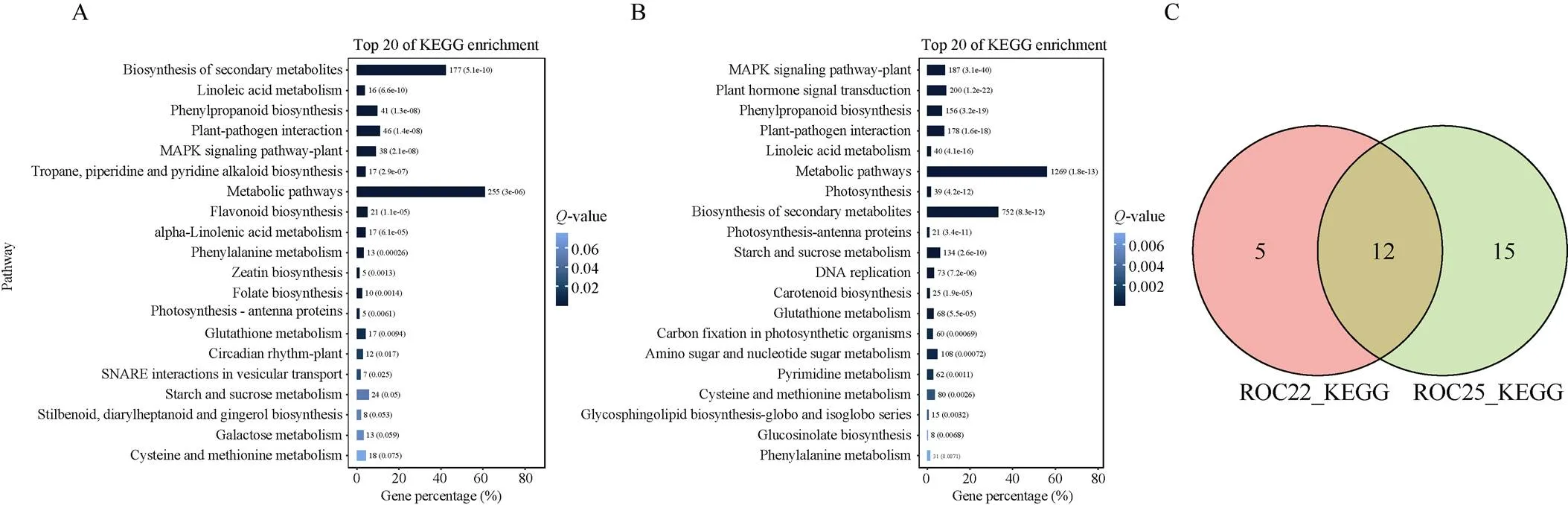
图8 ROC22和ROC25感染黑穗病菌后差异基因KEGG富集分析
A和B分别为ROC22和ROC25差异基因富集程度排名前20的KEGG通路。C为不同亲本的差异基因富集的KEGG通路比较分析。
A and B indicate TOP20 KEGG enrichment terms of differentially expressed transcripts in ROC22 and ROC25, respectively. C indicates comparison of KEGG pathways in different varieties.
2.8 植物MAPK信号通路在抗病反应中响应
前人研究表明植物-病原互作、植物MAPK信号通路、亚油酸代谢、苯丙素生物合成、次级代谢产物合成和淀粉和蔗糖代谢等在甘蔗抵御黑穗病菌侵染过程中起重要作用[16,19-20], 这些抗病通路均在抗感亲本中被显著富集, 表明它们是新台糖亲本对黑穗病菌的基础防御反应。植物MAPK信号通路在植物抗病过程中起中心作用, 它不仅介导病原相关分子模式和病原菌的效应因子而触发植物免疫, 还参与了植物激素(如JA、乙烯和ABA)信号通路和活性氧调节的植物防御反应[47-48]。在本研究中, 植物MAPK信号通路在抗感亲本中都得到显著富集, 尤其在ROC25中富集程度最高, 意味着植物MAPK信号通路在新台糖亲本与黑穗病的互作中扮演着重要的角色, 但目前植物MAPK信号通路在响应黑穗病侵染中的作用机制尚不清楚, 为此, 本研究中对植物MAPK信号通路中关键MAPK超家族基因在抗感亲本中的表达差异进行了分析。结果表明, MAPK超家族基因中(8个转录本)、(2个转录本)、(5个转录本)、(6个转录本)、(1个转录本)和(7个转录本)共6个基因的29个转录本在抗感亲本黑穗病菌侵染后表达发生变化。由表5可知, 其中仅3个基因(,和)的6个转录本在ROC22中有显著差异表达,而29个转录本均在ROC25中表达发生明显变化。在感病亲本ROC22中,、和在感染黑穗病后5 d时表达均发生上调, 在8 d和11 d时则没有显著表达差异。在抗病亲本ROC25中,、、、、和共6个基因在感染黑穗病菌后8 d时表达水平均显著提高, 而在11 d时仅和两个基因的6个转录本的表达发生下降。总的来说, MAPK超家族基因在甘蔗感染黑穗病后的主要表达方式为上调, 但不同亲本和不同侵染时段的表达特征不一致, 在抗病亲本中响应的MAPK超家族基因明显多于感病亲本, 参与响应的的转录本数目在抗感亲本表达较接近, 而、和仅在ROC25中表达发生变化。
2.9 抗病相关转录因子在抗病反应中响应
转录因子(transcription factor, TF)是控制基因表达的重要调控因子, 在植物发育、细胞周期、细胞信号转导和胁迫反应等过程发挥重要作用。本研究分析了主要的抗病相关TF家族基因(如、、和等)在抗感亲本感染黑穗病菌后的表达规律, 以了解甘蔗对黑穗病菌胁迫的调控机制。由表6可知, 接种黑穗病菌后, 感病亲本ROC22有7个、4个、12个和15个基因的表达水平发生变化, 其中差异表达基因比例(差异基因数目/总检测基因数目)较高的是和家族; 而抗病亲本ROC25则有23个、13个、16个和20个基因的表达水平发生变化, 其中和家族的差异表达基因比例较高。抗感亲本中共同差异基因较多的是和家族, 而不同差异基因较多的是和家族。从基因表达模式上看, 除抗病亲本中家族的上/下调基因比例接近外, 大部分转录因子在两亲本中呈上调表达。从时间上看, 感病亲本在接种黑穗病菌5 d时拥有最多的差异表达基因, 而抗病亲本在8 d和11 d时拥有更多的差异表达基因。综上, ROC22和ROC25亲本中众多WRKY、MYB、NAC和AP2/ERF-ERF等抗病相关转录因子响应了黑穗病菌胁迫, 但转录因子类型、数目以及时间表达特征在抗感亲本间存在明显差异, 这些分子表达差异可能影响亲本的生理、生化变化, 最终形成不同的抗性表型。
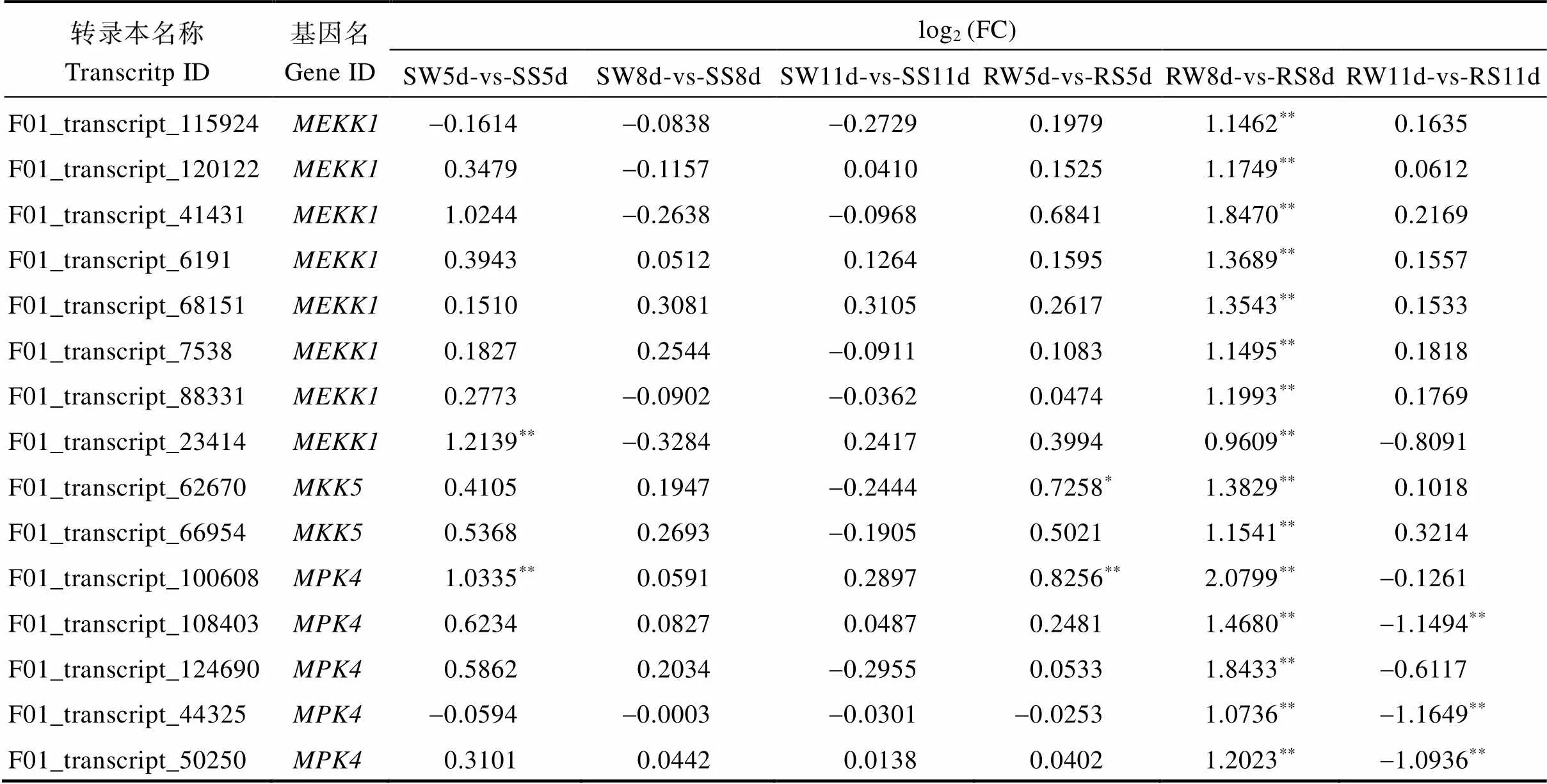
表5 ROC22 and ROC25感染黑穗病菌后差异表达的MAPK超级家族基因
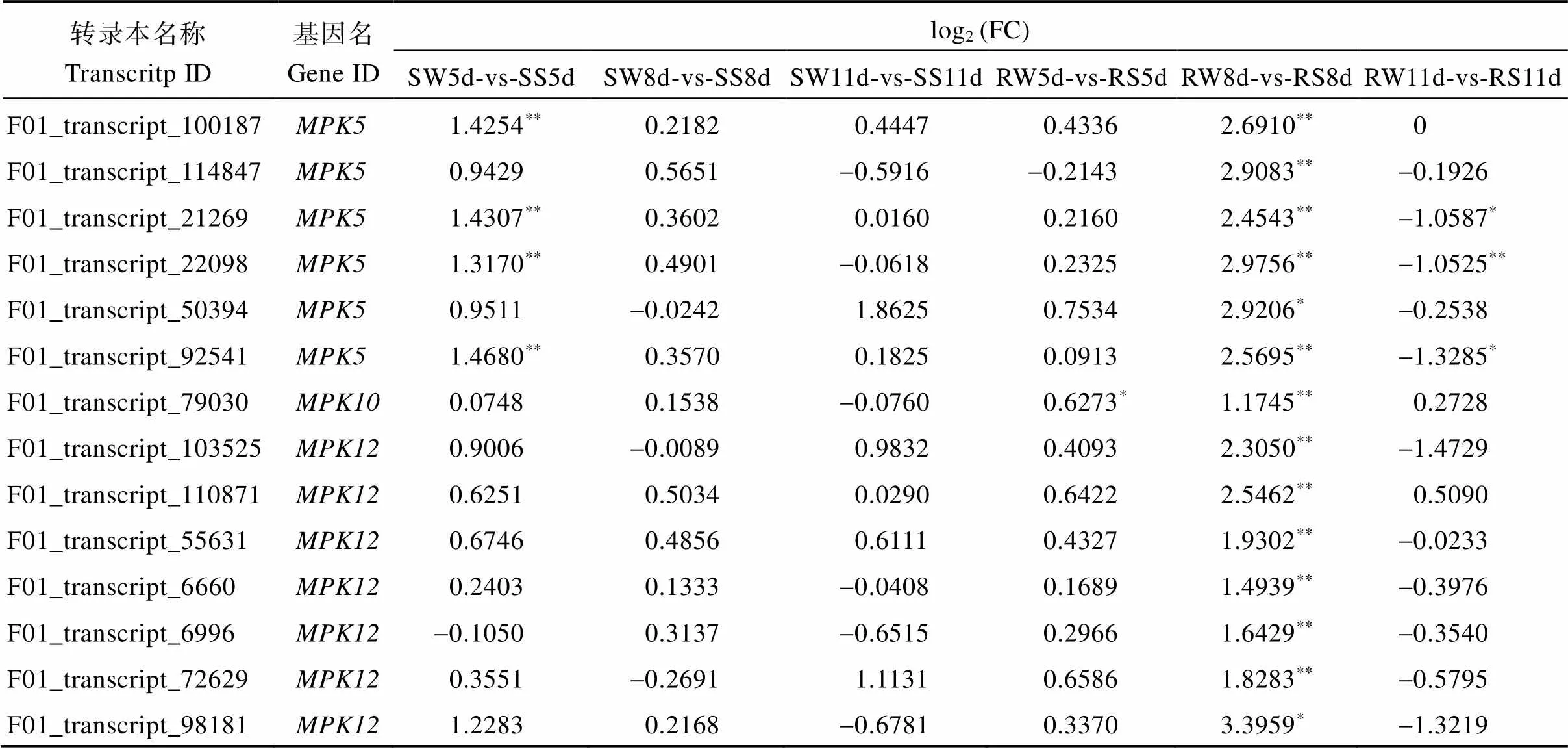
(续表5)
FC表示差异倍数。采用费希尔精确检验判断显著性水平,*和**分别表示错误发现率(FDR)的值小于0.05和0.01。
FC: fold changes. Significantly difference was determined by Fisher’s exact test.*and**represent that the false discovery rate (FDR)-value is less than 0.05 and 0.01, respectively.
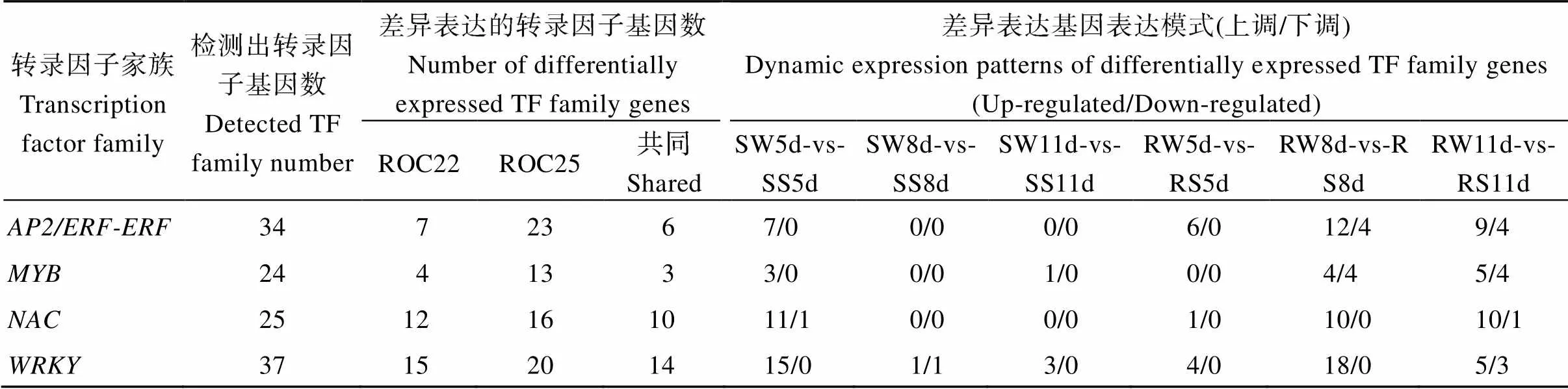
表6 ROC22 and ROC25感染黑穗病菌后抗病相关转录因子分析
3 讨论
新台糖亲本是目前甘蔗杂交育种重要的亲本, 已培育出我国新一代当家良种, 对其基因资源开展深入挖掘对于高效利用该类亲本具有重要意义。与二代测序相比, 三代测序的cDNA读长更长, 获得的长RNA序列更为准确, 因而更利于分析基因结构、可变剪切分析、LncRNA鉴定、融合基因分析等[29,49-50]。为了摸清该类亲本中的抗黑穗病基因资源, 本研究利用三代PacBio平台的长读长特点, 构建了我国重要甘蔗亲本ROC22和ROC25响应黑穗病侵染的全长转录组文库, 获得了79,885条转录本序列。在甘蔗与黑穗病菌互作后转录本分析中, 共鉴定到3692个可变剪接事件、1799个LncRNA和8002个转录因子, 这为后续转录组分析提供较好的遗传信息。进一步, 本研究结合二代Ilumina平台产生的对应样本的短片段转录组数据, 对三代全长转录组鉴定到的转录本进行分析, 使得定量更为准确, 突破了以往以基因为研究单元的转录组分析模式, 以更为接近生物学过程的转录本为研究单元对甘蔗与黑穗病互作过程进行了详细研究。
近年来, 利用转录组学和蛋白组学方法发现了一系列响应黑穗病菌侵染的差异基因和蛋白, 并揭示了甘蔗防御黑穗病菌侵染的重要代谢通路[16,20,51-52]。转录组学分析显示发现不同侵染阶段抗病亲本ROC25和感病亲本ROC22中筛选获得的差异基因数量和时间变化趋势明显不同。在感病亲本中, 侵染5 d时获得差异表达基因较多, 而抗病亲本在侵染8 d和11 d获得的的差异基因数目较多, 表明2个亲本对黑穗病菌胁迫积极响应时间不一致, 抗病亲本响应最激烈时段出现较晚, 持续反应时间更长。2个不同抗病亲本中基因响应模式差异可能与黑穗病菌定植过程有关, 这可能是由于抗病亲本阻碍黑穗病菌定植过程, 黑穗病菌进入抗病亲本茎尖分生组织的时间通常要晚于感病亲本[53-54]。另一方面, 在易感甘蔗中, 低效的防御过程和较弱的响应信号使黑穗病菌传播并损害植物[16,51], 在本研究中, 感病亲本中总体差异基因数目要低于抗病亲本, 这也反映抗病亲本对黑穗病侵染具有更积极和更长期的防御反应。通过对差异基因的GO和KEGG富集, 抗病亲本中差异表达基因共富集到的GO条目和KEGG通路均要多于感病亲本, 进一步佐证了抗病亲本中更多的生命过程和代谢途径积极参与对黑穗病的响应过程。以上结果与前人研究结果[16,20,51-52]基本一致, 表明苯丙素途径、次生代谢物生物合成、植物激素信号转导、植物-病原体相互作用等与抗性相关的代谢途径等重要代谢通路在新台糖亲本防御黑穗病过程中发挥了重要作用。本研究发现, 抗病亲本感染黑穗病菌后DNA复制、嘧啶代谢和卟啉代谢等DNA复制、修复相关的代谢途径通路受到显著影响, 表明这些生物学过程也参与调控甘蔗抗黑穗病反应。此外, 类胡萝卜素生物合成在抗病亲本中显著富集, 由于类胡萝卜素具有显著的抗氧化功能, 可减少细胞遗传物质和细胞膜的氧化损伤[55], 因而该通路激活可能有利于减少黑穗病菌引发的氧化应激反应, 进而提高甘蔗的免疫水平。
MAPK作为植物信号感受组分参与调控多种生命过程, MAPK信号途径通常依次激活3个相互连接的蛋白激酶MAPKKK-MAPKK-MAPK, 将信号依次级联放大并传递至细胞及核内[56]。它在植物中调控气孔、侧枝等的形成和生长发育, 参与细胞分裂, 应对各种生物和非生物胁迫及参与植物激素信号的转导调节等均发挥着至关重要的作用[48,57]。在拟南芥中, 至少有2个完整的MAPK级联, 即MEKK1- MKK1/MKK2-MPK4和MAPKKK3/MAPKKK5-MKK4/ MKK5-MPK3/MPK6级联, 并被报道参与了免疫应答的诱导。在单子叶植物水稻中,、、、、、、、和均被稻瘟病菌侵染诱导, 且其中有4个与寄主细胞死亡相关, 8个在防卫信号分子(如jasmonic acid, salicylic acid和ethylene)应答中出现差异性表达[58-64], 积极参与了水稻对病原菌的免疫反应。目前MAPK信号通路在甘蔗抵御黑穗病中作用研究报道较少。Wu等[65]通过Solexa测序鉴定了MAPK信号通路的3个基因、和在甘蔗感染黑穗病后表达发生上调, 其中在抗病品种Yacheng 05-179接种72 h后在表达显著上调, 而在易感的柳成03-182中的表达则不显著上调, 表明在甘蔗抵御黑穗病中起重要作用。本研究中, ROC22和ROC25在黑穗病菌侵染后中MAPK信号通路都被显著富集, 但在抗病亲本中具有更多差异表达的和的转录本, 且、和的表达水平仅在抗病亲本中发生变化, 暗示MAPK信号通路中差异响应基因与亲本抗病表型关联。进一步分析差异表达的家族基因在正常条件下的表达情况, 发现抗病品种(F01_tran- script_6996)在清水处理8 d时的表达水平是其在感病品种表达量的2.91倍(log2FC = 1.544,<0.01), 表明正常条件下抗病亲本具有较高的F01_transcript_6996表达水平, 且接种病菌后抗感病品种间F01_transcript_6996的表达差异进一步增大, 我们推测高水平的(F01_transcript_6996)可能有利于甘蔗抵御黑穗病。最新研究发现, 水稻可通过增强EREBP1转录因子活性, 调节基因表达水平, 从而提高植物的抗病水平[66-67];还可以通过激活转录因子来介导SA和JA诱导的防御反应[68-69]。本研究也发现,转录因子(F01_ transcript_24011, F01_transcript_40700, F01_transcript_ 94848, F01_transcript_119859)的表达也发生上调, 此外SA信号通路中转录因子(F01_transcript_ 35008, F01_transcript_37230, F01_transcript_43260, F01_transcript_47871, F01_transcript_94228, F01_ transcript_124105)和JA信号通路中转录因子(F01_transcript_3739, F01_transcript_4150, F01_ transcript_70898, F01_transcript_92492和F01_ transcript_98235)的表达也发生了变化, 因而也可能介导了植物激素SA和JA诱导的免疫反应。我们可以合理推测,家族基因在抗病亲本感染黑穗病菌后的免疫反应中发挥重要作用, 进一步的研究可以聚焦家族基因在甘蔗与黑穗病菌互作过程中的作用机制, 这将有助于解析甘蔗抗黑穗病的分子机制, 并为有效防治甘蔗黑穗病以及培育优良抗黑穗病品种提供理论依据。
WRKY、MYB、NAC和AP2/ERF-ERF是植物中主要的、功能多样化的转录因子家族, 在植物响应生物胁迫过程中发挥重要作用[70]。在植物对病原体的免疫反应中, WRKY参与了病原相关分子模式触发免疫(pattern-triggered immunity, PTI)、效应子触发免疫(effector-triggered immunity, ETI)和系统获得性抗性(systemic acquired resistance, SAR)等过程[71], NAC转录因子则影响了植物的ETI和超敏反应(hypersensitive response, HR)[72]。AP2/ERF转录因子可以通过JA、SA或ET介导的信号转导通路来积极调控植物的抗病性[73], 过表达[74]、[75]、[76]和[77]等均明显增强植物对病原菌的抗性水平。MYB转录因子也参与植物与病原菌互作过程, 它可以通过调控次级代谢产物合成、植物激素信号转导、防御基因表达等途径提高或降低植物对病原菌的抗性水平[78-81]。据Plant Transcription Factor Database统计, 甘蔗中共有73个、36个、44个和39个家族基因[82]。Que等[16]研究发现25个、18个和18个基因在甘蔗感染黑穗病菌后发生表达变化, 可能对甘蔗防御黑穗病菌起积极作用。最近, Agisha等[83]研究也揭示、、和等转录因子也参与了甘蔗与黑穗病菌互作过程。本研究通过全长转录组测序共获得7794个转录因子(占全部转录子的9.76%), 其中检测出、、和的基因数目分别占数据库各基因家族的94.9%、66.7%、56.8%和46.6% (表6)。抗感亲本中,和家族基因均积极响应了黑穗病菌胁迫, 暗示PTI、ETI、SAR和HR等免疫途径在抗感亲本均被激活, 以便于增强甘蔗对黑穗病菌的抗性。虽然响应的和基因种类在抗感品种间相似, 但基因表达水平差异也可能对亲本的抗性产生不同影响。例如,被认为与甘蔗抗黑穗病有关, 该基因在抗黑穗病品种中表达稳定, 而在感黑穗病品种中则表达下调[84]。在本研究中, 感染黑穗病菌5~8 d时,(F01_transcript_118129, 与的蛋白一致性达到99.5%)的表达水平在抗感亲本中均发生下降, 但相同条件下抗病亲本中的表达水平要高于感病品种, 因而推测高表达水平的有利于增加甘蔗对黑穗病菌的抗性。另一个可能调控甘蔗抗黑穗病的基因[23]在本研究中未被检测, 这可能由于样品类型及侵染时间不同。值得注意的是, 接种黑穗病菌后, 抗病亲本中激活或抑制的和的转录因子数量明显多于感病亲本, 暗示和家族基因在抗病亲本中发挥了更积极的调控作用。研究表明, ERF转录因子是ET和JA信号转导通路的调节因子, 抗病品种中甘蔗ERF1 (gi35045219)的表达受黑穗病菌诱导, 而在感病品种中其表达无差异[21]。本研究也发现(F01_transcript_116729) 在抗病亲本接种黑穗病菌5 d和8 d时, 表达水平分别上升了2.31倍和1.80倍, 而在感病亲本中表达水平无显著差异, 这与前人研究相近。除外,、、、、、、、、、、、和等多个家族基因仅在抗病亲本中发生变化, 也可能对亲本的抗黑穗病能力起积极作用。此外,、、、、、、、和等家族基因仅在抗病亲本中显示表达变化, 而转录因子则在感病亲本中出现转录差异, 它们也可能参与到甘蔗防御黑穗病菌过程。总的来说, 与感病亲本相比, 接种黑穗病菌后抗病品种显示出更多的抗病相关转录因子表达变化, 暗示了抗病亲本拥有更强的防御反应, 这些转录因子可能调控了多种生物学过程, 最终提高了甘蔗抗黑穗病水平。
4 结论
本研究以黑穗病菌侵染后新台糖亲本ROC22和ROC25为材料, 构建了全长转录组文库, 获得高质量转录本79,885条, 并对转录本进行了可变剪切分析、LncRNA鉴定、转录因子分析等, 从而完善了新台糖亲本基因组的注释信息。同时, 本研究结合Ilumina平台产生的二代转录组数据, 对三代全长转录组鉴定到的转录本进行了定量分析, 获得差异转录本并进行GO和KEGG富集分析, 获得了不同抗病亲本响应黑穗病菌的生物过程特征。进一步, 研究还揭示了植物MAPK信号通路、、、和等抗病相关转录因子在甘蔗抵御黑穗病菌过程中发挥重要作用, 这为深入解析甘蔗抗黑穗病的分子机制奠定了新基础。
[1] Magarey R C, Bull J I, Sheahan T, Denney D, Bruce R C. Yield losses caused by sugarcane smut in several crops in Queensland., 2010, 32: 347–354.
[2] Sundar A R, Barnabas E L, Malathi P, Viswanathan R, Sundar A R, Barnabas E L. A mini-review on smut disease of sugarcane caused by.,2012, 2014: 226.
[3] Magarey R C, Bull J I, Lonie K J, Piperidis G. The effect of smut resistance on disease incidence and severity under natural spread conditions., 2012, 34: 8.
[4] 王长秘, 李婕, 张荣跃, 王晓燕, 单红丽, 仓晓燕, 尹炯, 罗志明, 黄应昆. 甘蔗黑穗病研究进展. 中国糖料, 2021, 43(2): 65–70. Wang C M, Li J, Zhang R Y, Wang X Y, Shan H L, Cang X Y, Ying J, Luo Z M, Huang Y K. Research progress of sugarcane smut disease., 2021, 43(2): 65–70 (in Chinese with English abstract).
[5] Chao C, Hoy J, Saxton A, Martin FA. Heritability of resistance and repeatability of clone reactions to sugarcane smut in Louisiana., 1990, 80: 622–626.
[6] 许莉萍, 陈如凯. 甘蔗黑穗病及其抗病育种的现状与展望. 福建农业学报, 2000, 15(2): 26–31.Xu L P, Chen R K. Current status and prospects of smut and smut resistance breeding in sugarcane., 2000, 15(2): 26–31(in Chinese with English abstract).
[7] Alexander K C, Ramakrishnan K. Infection of the bud, establishment in the host and production of whips in sugarcane smut () of sugarcane., 1980, 17: 1452–1455.
[8] Aitken K S, Bhuiyan S, Berkman P J, Croft B, McNeil M. Investigation of the genetic mechanisms of resistance to smut in sugarcane., 2013, 28: 968–977.
[9] Marques J P R, Appezzato-da-Glória B, Piepenbring M, Massola Jr N S, Monteiro-Vitorello C B, Vieira M L C. Sugarcane smut: shedding light on the development of the whip-shaped sorus., 2017, 119: 815–827.
[10] Rajput M A, Rajput N A, Syed R N, Lodhi A M, Que Y X.Sugarcane smut: current knowledge and the way forward for management., 2021, 7: 1095.
[11] Dean L J. The effect of wounding and high-pressure spray inoculation on the smut reaction of sugarcane clones., 1982, 72: 1023–1025.
[12] Vitorello C B, Schaker PDC, Benevenuto J, Teixeira-Silva NS, Almeida SS. Progress in understanding fungal diseases affecting sugarcane: smut. In: Rott P, ed.Achieving Sustainable Cultivation of Sugarcane. Cambridge, UK:Burleigh Dodds Science Publishing, 2018. pp 221–243.
[13] Lloyd H L, Naidoo M. Chemical assay potentially suitable for determination of smut resistance of sugarcane cultivars., 1983, 67:1103–1105.
[14] Fontaniella B, Marquez A, Rodriguez CW, PinonD, Solas MT, Vicente C, Legaz ME. A role for sugarcane glycoproteins in the resistance of sugarcane to., 2002, 40:881–889.
[15] Millanes AM, Vicente C, Legaz ME. Sugarcane glycoproteins bind to surface, specific ligands and modify cytoskeleton arrangement ofteliospores., 2008, 3: 95–110.
[16] Que Y, Su Y, GuoJ, Wu Q, Xu L. A global view of transcriptome dynamics duringchallenge in sugarcane by RNA-seq., 2014, 9: e106476.
[17] Peters L P, Carvalho G, Vilhena M B, Creste S, Azevedo R A, Monteiro-Vitorello C B.Functional analysis of oxidative burst in sugarcane smut-resistant and-susceptible genotypes., 2017, 245: 749–764.
[18] Peters L P, Teixeira-Silva N S, BiniA P, Silva M M L, Moraes N, Crestana G S, Monteiro-Vitorello C B. Differential responses of genes and enzymes associated with ROS protective responses in the sugarcane smut fungus., 2020, 124: 1039–1051.
[19] Schaker P D, Palhares A C, Taniguti L M, Peters L P, Creste S, Aitken K S,Van Sluys M A, Kitajima J P, Vieira M L, Monteiro-Vitorello C B. RNAseq transcriptional profiling following whip development in sugarcane smut disease., 2016, 11: e0162237.
[20] Schaker P D, Peters L P, Cataldi T R, Labate C A, Caldana C, Monteiro-Vitorello C B. Metabolome dynamics of smutted sugarcane reveals mechanisms involved in disease progression and whip emission., 2017, 8: 882.
[21] Su Y, Xu L, Wang Z, Peng Q, Yang Y, Chen Y, Que Y. Comparative proteomics reveals that central metabolism changes are associated with resistance againstin sugarcane., 2016, 17:800.
[22] Bhuiyan S A, Magarey R C, McNeil M D, Aitken K S. Sugarcane smut, caused by, a major disease of sugarcane: acontemporary review., 2021, 111: 1905–1917.
[23] Wang D, Wang L, Su W, Ren Y, You C, Zhang C, Que Y, Su Y. A class III WRKY transcription factor in sugarcane was involved in biotic and abiotic stress responses., 2020, 10:20964.
[24] Huang N, Ling H, Zhang X, Mao H, Su Y, Su W, Liu F, Xu L, Chen R, Que Y. A small GTP-binding gene scran from sugarcane is involved in responses to various hormone stresses andchallenge., 2018, 20:669–680.
[25] Sun T, Cen G, You C, Lou W, Wang Z, Su W, Wang W, Li D, Que Y, Su Y., an allene oxide cyclase gene, confers defense response to biotic and abiotic stresses in sugarcane., 2020, 39: 1785–1801.
[26] Sun T, Liu F, Wang W, Wang L, Wang Z, Li J, Que Y, Xu L, Su Y. The role of sugarcane catalase genein the defense response to pathogen challenge and adversity stress., 2018, 19: 2686.
[27] Ren Y, Zou W, Feng J, Zhang C, Su W, Zhao Z, Wang D, Sun T, Wang W, Cen G, Que Y, Su Y. Characterization of the sugarcanegene family and the negative regulatory role ofin response to pathogen stress., 2022, 176: 114292.
[28] 吴才文,赵俊,赵培方,刘家勇,杨昆,夏红明,昝逢刚.几个新台糖甘蔗品种杂交育种潜力研究.西南农业学报, 2010,23: 1413–1417.Wu C W, Zhao J, Zhao P F, Liu J Y, Yang K, Xia H M, Zan F G. Research on breeding potential of several ROC varieties in sugarcane., 2010,23: 1413–1417 (in Chinese with English abstract).
[29] 赵理贤, 肖雪, 陈悦佳, 刘丹丹, 黄有总, 邹承武, 陈保善.33 份甘蔗种质资源的 ISSR 标记和遗传多样性.分子植物育种, 2022, https://kns.cnki.net/kcms/detail/46.1068.S.20220728.1128. 006.html.Zhao L, Xiao X, Chen Y, Liu D, Huang Y, Zou C, Chen B. ISSR marker polymorphism and genetic diversity of 33 sugarcane germplasm resources., 2022, https://kns.cnki. net/kcms/detail/46.1068.S.20220728.1128.006.html(in Chinese with English abstract).
[30] 邓海华, 张琼. 我国大陆近年育成甘蔗品种的亲本分析, 广东农业科学, 2006,12(2): 7–10. Deng HH, Zhang Q. Analysis on the parents of commercial varieties released in Mainland China in recent years., 2006, 12(2): 7–10 (in Chinese with English abstract).
[31] 刘新龙, 李旭娟, 刘洪博, 马丽, 徐超华, 范源洪. 云南甘蔗常用亲本资源遗传多样性的SSR分析. 植物遗传资源学报, 2015, 16:1212–1222. Liu X L, Li X J, Liu H B, Ma L, Xu C H, Fan Y H. Genetic diversity analysis of Yunnan commonly-used parents by using SSRmarker., 2015, 16:1212–1222 (in Chinese with English abstract).
[32] 沈万宽, 姜子德, 杨湛端, 刘睿, 陈健文, 邓海华. 甘蔗抗黑穗病的鉴定新方法及其品种抗性评价. 华中农业大学学报, 2014, 33(2): 51–56.Shen W K, Jiang Z D, Yang Z D, Liu R, Chen J W, Deng H H. New resistance identification method and resistance evaluation of sugarcane varieties to smut disease., 2014, 33(2): 51–56 (in Chinese with English abstract).
[33] 阙友雄,许莉萍,林剑伟,陈天生,陈如凯,李依龙. 甘蔗品种黑穗病抗性评价体系的建立(英文). 植物遗传资源学报,2006,7:18–23. Que Y X, Xu L P, Lin J W, Chen T S, Chen R K, Li Y L. Establishment of evaluation system of smut resistance for sugarcane varieties., 2006,7:18–23.
[34] 苏亚春. 甘蔗应答黑穗病菌侵染的转录组与蛋白组研究及抗性相关基因挖掘.福建农林大学博士学位论文,福建福州, 2014.Su Y C. Transcriptomics and Proteomics of Sugarcane Response toInfection and Mining of Resistance-related Genes. PhD Dissertation of Fujian Agriculture and Forestry University, Fuzhou, Fujian, China, 2014 (in Chinese with English abstract).
[35] Thomas S, Underwood JG, Tseng E, Holloway AK. Long-read sequencing of chicken transcripts and identification of new transcript isoforms., 2014, 9: e94650.
[36] Hackl T, Hedrich R, Schultz J, Förster F. Proovread: large-scale high-accuracy PacBio correction through iterative short read consensus., 2014, 30: 3004–3011.
[37] Li W Z, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences., 2006, 22: 1658–1659.
[38] SimãoF A, WaterhouseR M,Ioannidis P, KriventsevaE V, Zdobnov E M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs., 2015, 31: 3210–3212.
[39] Liu X, Mei W, Soltis P S, Soltis D E, Barbazuk W B. Detecting alternatively spliced transcript isoforms from single-molecule long-read sequences without a reference genome., 2017, 17: 1243–1256.
[40] Kong L, Zhang Y, Ye Z Q, Liu X Q, ZhaoS Q, Wei L, Gao G. CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine., 2007, 35: 345–349.
[41] Wang L, Park H J, Dasari S, Wang S, Kocher J P, Li W. CPAT: Coding-Potential Assessment Tool using an alignment-free logistic regression model., 2013, 41: e74.
[42] Zheng Y, Jiao C, Sun H, Rosli HG, Pombo MA, Zhang P, Banf M, Dai X, Martin GB, Giovannoni JJ, Zhao PX, Rhee SY, Fei Z. iTAK: a program for genome-wide prediction and classification of plant transcription factors, transcriptional regulators, and protein kinases., 2016, 9:1667–1670.
[43] Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras, T R. STAR: ultrafast universal RNA-seq aligner., 2013,29: 15–21.
[44] Li B, Dewey C N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome., 2011, 12: 323.
[45] Anders S, Huber W. Differential expression analysis for sequence count data., 2010, 11:R106.
[46] 阙友雄, 许莉萍, 徐景升, 张积森, 张木清, 陈如凯. 甘蔗基因表达定量PCR分析中内参基因的选择. 热带作物学报, 2009, 30(3): 274–278. Que Y X, Xu L P, Xu J S, Zhang J S, Zhang M Q, Chen R K. Selection of control genes in real-time qPCR analysis of gene expression in sugarcane., 2009, 30(3): 274–278 (in Chinese with English abstract).
[47] Galletti R, Ferrari S, De Lorenzo G.MPK3 and MPK6 play different roles in basal and oligogalacturonide- or flagellin-induced resistance against., 2011, 157: 804–814.
[48] 杨洪强,接玉玲.植物MAPK及其在病原信号传递中的作用.植物病理学报,2003, 33:8–13.Yang H Q, Jie Y L. The plant MAPK and its function in pathogen signaling cascades., 2003, 33:8–13 (in Chinese with English abstract).
[49] Gordon S P, Tseng E, Salamov A, Zhang J, Meng X, Zhao Z, Kang D, Underwood J, Grigoriev I V, Figueroa M, Schilling J S, Chen F, Wang Z. Widespread polycistronic transcripts in fungi revealed by Single-Molecule mRNA Sequencing., 2015, 10: e0132628.
[50] Abdel-Ghany S E, Hamilton M, Jacobi J L, Ngam P, Devitt N, Schilkey F, Ben-Hur A, Reddy A S. A survey of the sorghum transcriptome using single-molecule long reads., 2016, 7:11706.
[51] Vicente C, Legaz ME, Sánchez-Elordi E. Physiological basisof smut infectivity in the early stagesof sugar cane colonization., 2021, 7: 44.
[52] Peters L P, Teixeira-Silva N S, Bini A P, Silva M M L, Moraes N, Crestana G S, Creste S, Azevedo R A, Carvalho G, Monteiro-Vitorello C B.Differential responses of genes and enzymes associated with ROS protective responses in the sugarcane smut fungus., 2020, 124: 1039–1051.
[53] Ferreira S A, Comstock J C,Wu K K. Evaluating sugarcane for smut resistance., 1980, 17: 1463–1476.
[54] Marques JPR, Hoy JW,Appezzato-da-Glória B, Viveros AFG, Vieira MLC, Baisakh N. Sugarcane cell wall-associated defense responses to infection by., 2018, 9: 698.
[55] Dhar M K, Mishra S, Bhat A, ChibS, Kaul S. Plant carotenoid cleavage oxygenases: structure-function relationships and role in development and metabolism., 2020, 19:37.
[56] Widmann C, Gibson S, Jarpe MB, Johnson GL. Mitogen- activated protein kinase: conservation of a three-kinase module from yeast to human., 1999, 79: 143–180.
[57] Mao G, Meng X, Liu Y, Zheng Z, Chen Z, Zhang S. Phosphorylation of a WRKY transcription factor by two pathogen-responsive MAPKs drives phytoalexin biosynthesis in.,2011, 23: 1639–1653.
[58] Fu SF, Chou W C, Huang D D, Huang H J.Transcriptional regulation of a rice mitogen-activated protein kinase gene,, in response to environmental stresses., 2002, 43: 958–963.
[59] AgrawalG K, Agrawal S K, Shibato J, Iwahashi H, Rakwal R. Novel rice MAP kinases OsMSRMK3 and OsWJUMK1 involved in encountering diverse environmental stresses and developmental regulation., 2003, 300: 775–783.
[60] Zhang H, Liu Y, Wen F, Yao D, Wang L, Guo J, Ni L, Zhang A, Tan M, Jiang M. A novel rice C2H2-type zinc finger protein, ZFP36, is a key player involved in abscisic acid-induced antioxidant defence and oxidative stress tolerance in rice., 2014, 65: 5795–5809.
[61] Kishi-Kaboshi M, Okada K, Kurimoto L, Murakami S, Umezawa T, Shibuya N, Yamane H, Miyao A, Takatsuji H, Takahashi A, Hirochika H. A rice fungal MAMP-responsive MAPK cascade regulates metabolic flow to antimicrobial metabolite synthesis.,2010, 63: 599–612.
[62] Song F, Goodman R M., a rice MAP kinase gene involved in disease resistance responses., 2002, 215:997–1005.
[63] Wang Q, Li J, Hu L, ZhangT, Zhang G, Lou Y.positively regulates the JA signaling pathway and plant resistance to a chewing herbivore in rice., 2013, 32: 1075–1084.
[64] Reyna N S, Yang Y. Molecular analysis of the rice MAP kinase gene family in relation toinfection., 2006, 19: 530–540.
[65] Wu Q, Xu L, Guo J, Su Y, Que Y. Transcriptome profile analysis of sugarcane responses toinfection using Solexa sequencing technology., 2013, 2013: 9.
[66] Nakagami H, Pitzschke A, Hirt H. Emerging MAP kinase pathways in plant stress signalling., 2005, 10: 339–346.
[67] Cheong Y H, Moon B C, Kim J K, Kim C Y, Kim M C, Kim I H, Park C Y, Kim J C, Park B O, Koo S C, Yoon H W, Chung W S, Lim C O, Lee S Y, Cho M J. BWMK1, a rice mitogen-activated protein kinase, locates in the nucleus and mediates pathogenesis-related gene expression by activation of a transcription factor., 2003, 132: 1961–1972.
[68] Koo S C, Moon BC, Kim J K, Kim C Y, Sung S J, Kim M C, ChoM J, Cheong Y H.mediates SA-dependent defense responses by activating the transcription factor., 2009, 387: 365–370.
[69] Ning J, Yuan B, Xie K B, Hu H H, Wu C Q, Xiong L Z. Isolation and identification of SA and JA inducible protein kinase genein rice.,2006, 33: 625–633.
[70] Javed T, Shabbir R, Ali A, Afzal I, Zaheer U, Gao S J. Transcription factors in plant stress responses: challenges and potential for sugarcane improvement., 2020, 9: 491.
[71] Chen F, Hu Y, Vannozzi A, Wu K, Cai H, Qin Y, Mullis A, Lin Z, Zhang L. The WRKY transcription factor family in model plants and crops., 2017, 36: 311–335.
[72] Yuan X, Wang H, Cai J, Li D, Song F. NAC transcription factors in plant immunity., 2019, 1: 3.
[73] Feng K, Hou X L, Xing G M, Liu J X, Duan A Q, Xu Z S, Li M Y,Zhuang J, Xiong A S. Advances in AP2/ERF super-family transcription factors in plant., 2020, 40: 750–776.
[74] Zang Z, Lyu Y, Liu S, Yang W, Ci J, Ren X, Wang Z, Wu H, Ma W, Jiang L, Yang W. A novel ERF transcription factor,, positively regulates maize resistance to,2020,11: 850.
[75] Zhao Y, Chang X, Qi D, Dong L, Wang G, Fan S, Jiang L, Cheng Q, ChenX, Han D, Xu P, Zhang S. A novel soybean ERF transcription factor,, increases resistance toinfection in soybean., 2017, 8: 299.
[76] Hawku M D, Goher F, Islam M A, Guo J, He F, Bai X, Yuan P, Kang Z, Guo J., an AP2/ERF transcription factor, is positively involved in wheat resistance tof. sp.., 2021, 22: 2080.
[77] Ji S, Liu Z, Wang Y. Trichoderma-induced ethylene responsive factormediates defense responses in.,2021, 12: 708010.
[78] He J, Liu Y, Yuan D, Duan M, Liu Y, Shen Z, Yang C, Qiu Z, Liu D, Wen P, Huang J, Fan D, Xiao S, Xin Y, Chen X, Jiang L, Wang H, Yuan L, Wan J. An R2R3 MYB transcription factor confers brown planthopper resistance by regulating the phenylalanine ammonia-lyase pathway in rice., 2020, 117: 271–277.
[79] Zhu X, Li X, He Q, Guo D, Liu C, Cao J, Wu Z, Kang Z, WangX.: a novel R2R3-MYB transcription factor involved in wheat defense against stripe rust.,2021, 12: 783388.
[80] Gu K D, Zhang Q Y, Yu J Q, Wang J H, Zhang F J, Wang C K, Zhao Y W, Sun C H, You C X, Hu D G, Hao YJ. R2R3-MYB transcription factorconfers increased resistance to the fungal pathogenin applesthe salicylic acid pathway., 2021, 69: 447–458.
[81] Shan T, Rong W, Xu H, Du L, Liu X, Zhang Z. The wheat R2R3-MYB transcription factorparticipates in resistance response against the pathogeninfection through regulating defense genes., 2016, 6: 28777.
[82] Jin JP, Tian F, Yang DC, Meng YQ, Kong L, Luo JC, Gao G.PlantTFDB 4.0: toward a central hub for transcription factors and regulatory interactions in plants., 2017, 45:D1040–D1045.
[83] Agisha VN, Ashwin NMR, Vinodhini RT, Nalayeni K, Ramesh S A, Malathi P, Viswanathan R. Transcriptome analysis of sugarcane reveals differential switching of major defense signaling pathways in response toisolates with varying virulent attributes., 2022, 13:969826.
[84] Wang L, Liu F, Zhang X, Wang W, Sun T, Chen Y, Dai M, Yu S, Xu L, Su Y, Que Y. Expression characteristics and functional analysis of thegene from sugarcane., 2018, 19: 4059.
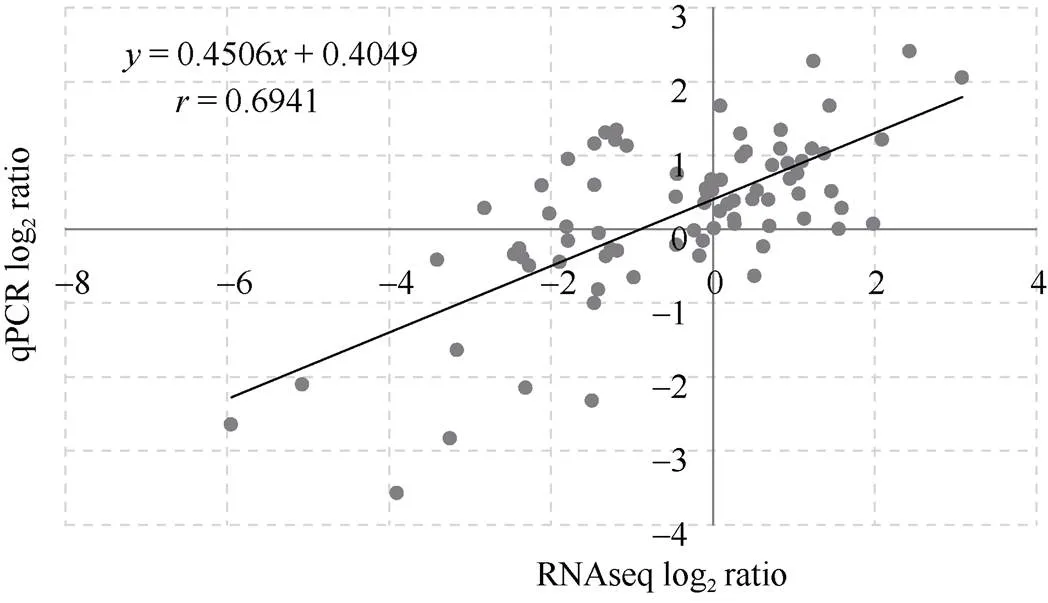
附图1 RNA-Seq和qRT-PCR相关性
Fig. S1 Correlations between RNA-Seq and qRT-PCR
Comparative transcriptome analysis of elite ‘ROC’ sugarcane parents for exploring genes involved ininfection by using Illumina- and SMRT-based RNA-seq
HU Xin, LUO Zheng-Ying, LI Chun-Jia, WU Zhuan-Di, LI Xu-Juan, and LIU Xin-Long*
Sugarcane Research Institute, Yunnan Academy of Agricultural Sciences / Yunnan Key Laboratory of Sugarcane Genetic Improvement / Key Laboratory of Sugarcane Biology and Genetic Breeding, Ministry of Agriculture and Rural Affairs (Yunnan), Kaiyuan 661699, Yunnan, China
Sugarcane smut, caused by the fungus, is the most challenging disease of sugarcane, causing significant losses in cane yield. There is a dearth of information on smut resistance mechanism in elite parents for the development of smut-resistant varieties. In the present study, we adopted joint Illumina- and Single Molecule Real-Time (SMRT)-based RNA-seq analysis to identify transcript expression in an smut-resistant and -susceptible parents (ROC25 and ROC22) infected with. A total of 79,885 high-quality transcripts was obtained, including 60,115 open reading frames, 3692 alternate splicing isoforms, 1799 long non-coding RNAs, 29,139 simple sequence repeats, and 7794 transcription factors. About 92.72% of the total transcripts were annotated, which should have increased the available data amount for transcriptome profile analysis. There were 2033 and 9716 differentially expressed transcripts (DETs) in ROC22 and ROC25, respectively. The analyses of GO and KEGGenrichment showed that more GO terms and KEGG pathways were observedin ROC25 than ROC22. It was found that MAPK signalling pathway-plant,phenylpropanoid biosynthesis, plant-pathogen interaction,linoleic acid metabolism, and starch and sucrose metabolism were enriched both in resistant and susceptible parents. In addition, MAPK superfamily genes were differentially regulated in different parents, more DETs ofandwere detected in resistant parent, and the relative expression levels of,,andgenes were specifically altered in resistant parent. It suggested that MAPK superfamily genes might play the important roles in the regulation of sugarcane response toinfection. Moreover, lots of transcription factors (TFs) associated with plant disease resistance were found to respond toinfection in both ROC22 and ROC25 parents, including WRKY, MYB, NAC, and AP2/ERF-ERF. Majority of the TFs were up-regulated. Compared to the susceptible ROC22 parent, the numberof activated transcription factors in the resistant ROC25 parent was higher, indicating that these extra TFs might have positive effects in the defense against. This study provides a comprehensive set of reference transcripts for sugarcane and thus increases our understanding on the interactions between sugarcane and,which should be helpful inguiding on exploitation and utilization of smut-resistance gene resources.
sugarcane;; full-length transcriptomes; RNA-seq; ROC
2022-10-12;
2023-02-21;
2023-03-10.
10.3724/SP.J.1006.2023.24228
通信作者(Corresponding author):刘新龙, E-mail: lxlgood868@163.com
E-mail: sugarhuxin@163.com
本研究由云南基础研究计划项目(2019FA016), 云南基础研究计划项目(202201AU070200)和云南省种子种业联合实验室项目(202205AR070001-09)资助。
This study was supported by the Yunnan Fundamental Research Projects (2019FA016), the Yunnan Fundamental Research Projects (202201AU070200), and the Yunnan Seeds and Seed Industry Joint Laboratory Project (202205AR070001-09).
URL: https://kns.cnki.net/kcms/detail/11.1809.S.20230309.1742.008.html
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
