β-地中海贫血合并肝血色病漏诊遗传性球形红细胞增多症一例
2023-07-20向丹丹鲁艳军曾志林杨道锋倪明
向丹丹 鲁艳军 曾志林 杨道锋 倪明
患者,男,42岁,因“皮肤巩膜黄染30余年加重1年”于2017年8月16日入院。患者30余年前无明显诱因出现皮肤巩膜黄染和尿色加深,不伴发热、乏力、纳差、皮肤瘙痒和大便颜色变浅,未诊治。近1年来自觉症状较前加重,为确诊来我院。既往史:否认病毒性肝炎、饮酒及输血史。其儿子亦自幼皮肤巩膜黄染。体格检查:T 36.5 ℃,R 19次/分,P 76次/分,Bp 120/60 mmHg,贫血貌,皮肤巩膜黄染,无肝掌及蜘蛛痣,心肺听诊无异常,腹平软,肝脏肋下未触及,脾脏肋下2 cm,质软。实验室检查:血常规:WBC计数3.42×109/L(3.50~9.50×109/L,括号内为正常参考值范围,以下相同),红细胞计数3.16×1012/L(4.30~5.80×1012/L),血红蛋白84 g/L(130~175 g/L),平均血红蛋白含量26.6 pg(27.0~34.0 pg),平均血红蛋白浓度311 g/L(316~354 g/L),红细胞分布宽度19.4(<14.9),红细胞分布宽度SD 60 fL(39~46 fL),PLT计数108×109/L(125~350×109/L),网织红细胞计数0.132×1012/L(0.025~0.075×1012/L),平均红细胞体积正常。肝功能:总胆红素102.3 μmol/L(≤21.0 μmol/L),直接胆红素9.9 μmol/L(≤5.0 μmol/L),间接胆红素92.4 μmol/L(≤13.3 μmol/L),AST、ALT结果均正常。实验室检查结果提示溶血性贫血。溶血性贫血筛查结果:游离血红蛋白117 mg/L(<40 mg/L),结合珠蛋白<0.06 g/L(0.36~1.95 g/L)。红细胞寿命12天(≥75天);红细胞形态未见明显异常;盐水渗透脆性试验结果正常;Coomb’s试验阴性;CD55、CD59表达均未见异常;葡萄糖-6-磷酸脱氢酶(G6PD)活性正常。骨髓细胞学和骨髓活检结果:红细胞系造血系统增生明显-极度活跃,以中晚幼红细胞为主,形态未见明显异常,成熟红细胞大小不等,可见泪滴型红细胞。血红蛋白电泳结果显示血红蛋白A2(3.5%)和血红蛋白F(19%)水平均升高,提示β地中海贫血。β地中海贫血常见基因突变检测结果示IVS-Ⅱ-654杂合突变。患者腹部MRI检查示肝脏T1和T2信号显著降低(图1),提示肝血色病。铁代谢结果:血清铁34.83 μmol/L(6.60~26.00 μmol/L),不饱和铁结合力6.9 μmol/L(24.2~70.1 μmol/L),转铁蛋白1.83 g/L(2.00~3.60 μmol/L),可溶性转铁蛋白受体13.14 mg/L(1.90~4.40 mg/L),铁蛋白798.3 μg/L(15.0~150.0 μg/L),转铁蛋白饱和度83.5%(16.0%~45.0%),总铁结合力正常。铁代谢结果示铁超载。为排除遗传性血色病,对患者进行了全外显子测序,结果未发现遗传性血色病相关基因突变,而检出遗传性球形红细胞增多症相关的SLC4A1杂合基因突变c.2102G>A(图2A)和Gilbert综合征相关的UGT1A1*1/28杂合突变(图2B)。根据上述结果,最终诊断:遗传性球形红细胞增多症并β-地中海贫血,继发肝血色病;Gilbert综合征。患者儿子血常规结果:红细胞计数6.45×109/L,平均红细胞体积62.9 fL(82.0~100.0 fL),平均血红蛋白含量20.6 pg,红细胞分布宽度16.6,红细胞分布宽度SD 36.0,网织红细胞计数0.100×1012/L,血红蛋白、平均血红蛋白浓度、PLT计数均正常。肝功能:总胆红素50 μmol/L,直接胆红素8.0 μmol/L,间接胆红素42.0 μmol/L,AST、ALT均正常。铁代谢结果:可溶性转铁蛋白受体5.76 mg/L,转铁蛋白饱和度46.3%,血清铁、不饱和铁结合力、转铁蛋白、铁蛋白、总铁结合力均正常。血红蛋白电泳结果提示血红蛋白A2(4.8%)和血红蛋白F(4.9%)水平升高。β地中海贫血常见基因突变检测显示IVS-Ⅱ-654杂合突变。患者儿子的全外显子测序结果与患者本人完全相同。但其儿子铁超载程度较轻,腹部影像学检查未提示肝血色病表现。确诊后患者连续7天口服苯巴比妥每次30 mg、每日3次,间接胆红素没有明显下降;建议患者驱铁治疗,但因其经济原因放弃,至目前病情尚稳定。
讨 论
血色病是指由各种原因造成铁在实质器官中过度沉积[1],可分为遗传性和继发性。遗传性血色病是一组遗传性铁超载疾病,临床表现包括肝功能不全、糖尿病和心肌病[2]。继发性血色病是由于反复溶血、长期输血或过量饮酒导致网状内皮系统铁蓄积能力饱和,从而在实质细胞内产生铁沉积。本例患者铁代谢检测指标和腹部影像学检查结果与肝血色病一致,但没有肝功能不全、糖尿病或心肌病的临床表现,且全外显子测序没有发现遗传性血色病相关基因突变,因此考虑患者体内铁超载是由于反复溶血所致。
β-地中海贫血是由β-珠蛋白基因突变引起的β-珠蛋白水平不足的一组血红蛋白病,可分为轻型、中间型和重型[3-4]。轻型β-地中海贫血的基因特征主要是存在单位点的杂合突变,临床症状轻微。IVS-Ⅱ-654(C→T)突变在我国β地中海贫血患者中很常见[5],在该类患者中可检测到超过15%的正常β珠蛋白编码mRNA和正常的珠蛋白结构[6]。因此,本例患者β-地中海贫血为轻型,难以解释其临床的表现。遗传性球形红细胞增多症是由于细胞骨架相关基因突变引起红细胞膜结构的缺陷。据估计,遗传性球形红细胞增多症在我国男性中的患病率为1.27/10万,女性为1.49/10万[7],远低于北美和北欧[8-9]。遗传性球形红细胞增多症的诊断通常基于临床特征、家族史、体格检查(脾脏增大、黄疸)和实验室数据(全血细胞计数、形态学和网织红细胞计数)[10],基因诊断技术通常不被推荐为常规诊断方法。在非典型病例,尤其是伴有地中海贫血的病例中,因前者主要导致红细胞表面/体积比降低,而后者导致表面/体积比增加[11],两种疾病共存时可导致红细胞表面/体积比接近正常,客观上减轻了溶血的程度,易引起漏诊。两种疾病共存时,患者血常规中平均红细胞体积、平均血红蛋白含量和平均血红蛋白浓度显著降低,红细胞分布宽度显著高于正常对照组[12]。本例患者及其儿子的血常规结果均不典型,二者外周血细胞形态检测没有发现明显异常,虽然患者骨髓细胞学中发现有泪滴型红细胞,但由于缺乏遗传性球形红细胞增多症的其他特征表现,因此被漏诊。在类似病例中,基因诊断技术是一种有价值的诊断工具。与遗传性球形红细胞增多症相关的基因缺陷主要包括SPTA1、SPTB、ANK1、SLC4A1和EPB42。美国和欧洲ANK1突变占40%~65%;日本40%~50%的患者携带EPB42突变[8]。我国大多数患者存在SLC4A1突变[13]。SLC4A1基因编码的蛋白为带3蛋白,介导红细胞膜的阴离子的交换,其突变的主要类型是错义和移码突变,大多数集中在外显子区域[11]。本例患者的杂合基因突变c.2102G>A尚未见报道。SLC4A1杂合突变所导致的病情较轻,因此确诊率较低[14]。

图1 2017年8月12日患者腹部MRI检查结果
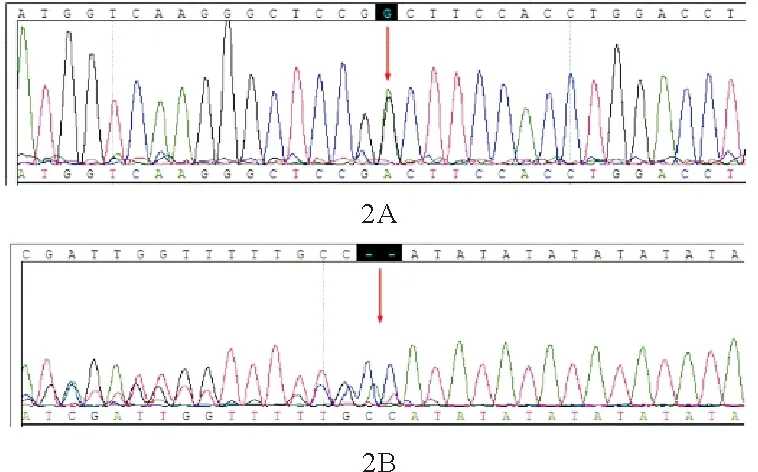
图2 2017年8月18日患者基因检测结果(A:SLC4A1杂合基因突变c.2102G>A;B:UGT1A1*1/28杂合突变)
Gilbert综合征是由于UGT1A1突变抑制了UGT1的合成和活性,从而导致间接胆红素升高。UGT1A1*28纯合突变最常见[15],其导致的血清胆红素水平较其他突变更高[16]。有研究结果提示,合并该突变的β-地中海贫血患者胆红素水平明显升高[17],本例患者是UGT1A1*1/28杂合突变,且口服苯巴比妥治疗无效,因此患者的间接胆红素水平升高并非UGT1A1功能下降引起。
目前尚未见遗传性球形红细胞增多症合并β-地中海贫血和Gilbert综合征的报道,也不清楚这3种遗传病之间是否存在相互作用。该病例提示对于地中海贫血患者需警惕合并遗传性球形红细胞增多症的可能,基因诊断技术对避免遗传性球形红细胞增多症的漏诊具有一定意义。
