临床试验设计的新型方案概述
2023-07-11王瑞平李斌
王瑞平 李斌
摘 要 近年来,随着分子生物学和临床研究技术的进步,疾病的诊疗进入精准医学时代。诊疗技术的进步为广大病患带来疾病治愈福音的同时,新型药物和治疗技术的研发也对临床试验设计提出了更高的要求,传统的临床试验设计方案已无法满足新型诊疗技术研发需求。因此,在临床研究方法学领域出现了一些新型设计,促使临床试验设计逐步向高效和精准的方向发展。本文介绍“无缝设计”“篮式设计”“伞式设计”“主方案设计”“富集设计”“生物标志物分层设计”等新型临床试验设计方案,为研究者今后开展临床研究提供参考。
关键词 临床试验设计 无缝设计 伞式设计 篮式设计 主方案设计 富集设计 生物标志物分层设计
中图分类号:R-3 文献标志码:C 文章编号:1006-1533(2023)09-0053-05
引用本文 王瑞平, 李斌. 临床试验设计的新型方案概述[J]. 上海医药, 2023, 44(9): 53-57.
基金项目:上海市卫生健康委员会卫生行业临床研究专项(202240371);上海申康医院发展中第二轮促进市级医院临床技能与临床创新三年行动计划——研究型医师创新转化能力培训项目(SHDC2022CRS053);上海市皮肤病医院引进人才科研基金项目(2021KYQD01);上海人才发展基金资助项目(2021SHRCFZ01);上海市医院协会医院管理研究基金项目(X2022117);中国医疗装备协会科研项目(LCIIT-2022-03);上海麦色IIT临床研究项目(LCIIT-2022-09);上海申康医院发展中心(局级)管理课题(2021SKMR-18)
Novel protocols for clinical trial design
WANG Ruiping, LI Bin
(Clinical Research & Innovation Center, Shanghai Skin Disease Hospital, Shanghai 200443, China)
ABSTRACT In recent years, with the advancement of molecular biology and clinical research technology, the treatment of diseases has entered the era of precision medicine. While these advances in treatment technology have brought cures to patients, the development of new drugs and therapeutic technologies has also placed greater demands on clinical trial design, and traditional clinical trial design protocols are no longer able to meet the demands of new treatment technology development. As a result, new designs have emerged in the field of clinical research methodology, leading to the development of efficient and precise clinical trial designs. The new clinical trial design options such as “seamless design”, “basket design”, “umbrella design”, “master design”, “enrichment design” and “biomarker stratification design” were introduced, so as to provide reference for researchers to conduct clinical studies in the future.
KEY WORDS clinical trial design; seamless design; umbrella design; basket design; master design; enrichment design; biomarker stratification design
隨着分子生物学、医学检测技术和临床研究技术的进步,针对危重疑难疾病的临床诊断和药物研发飞速发展。近年来,疾病的诊断和治疗逐步走进精准医学时代,例如肿瘤药物治疗,已从过去相对单一粗放式的放疗或化疗,逐步走向靶向治疗、免疫治疗等共存时代。诊疗技术的进步为广大患者带来疾病治愈福音的同时,新型药物和治疗技术的研发也对临床试验设计提出了更高的要求,传统的临床试验研究设计方案已无法满足新型诊疗技术研发需求[1]。因此,近年来在临床研究方法学领域出现了一些新型设计,促使临床试验设计逐步向高效和精准的方向发展。本文以新型药物研发为切入口,介绍近年来进入临床研究的“无缝设计”“篮式设计”“伞式设计”“主方案设计”“富集设计”“生物标志物分层设计”等新型临床试验设计方案,为研究者今后开展临床研究提供参考。
1 Ⅰ期临床试验方案
传统意义上来讲,Ⅰ期临床药物试验主要目的是确认最大耐受剂量或最佳生物效应剂量,同时为后续的Ⅱ期临床研究推荐剂量。近年来,为快速达成药物研发的主要目的,越来越多的新型Ⅰ期临床试验设计方案问世。Ⅰ期药物临床试验的研究方法可分为3大类:①基于规则/算法的设计,包括传统的“3+3”药物临床试验设计、加速滴定设计及药理学指导剂量爬坡方法设计等;②基于模型的设计,包括连续重评估方法设计、控制过量用药的剂量递增方法设计等;③基于模型辅助的设计,包括贝叶斯最优区间设计、改进的毒性概率区间设计、mTPI-2设计、Keyboard设计、“i3+3”设计等[2]。传统的“3+3”药物临床试验设计是在假设药物效应和毒性随剂量增加而增加的条件下进行剂量增减。由于不能按临床需要变化、获得最大耐受剂量(maximum tolerated dose, MTD)准确偏差值,可能会导致在指导后续Ⅱ期临床试验时,又由于剂量不足而不能显现应有的疗效。为克服传统的“3+3”药物临床试验缺陷,便产生了加速滴定设计,其主要特点为快速的初始剂量递增并允许对同一受试者进行剂量递增,有助于减少接受低剂量治疗的受试者数量并能加快研究进度。药理引导的剂量递增设计可实时测量每位患者的药动学数据以确定随后的剂量水平,只要血药浓度-时间的曲线下面积未达预先定义水平,就按每剂量一个患者进行剂量爬坡,剂量增量通常为100%,而一旦达到目标血药浓度-时间曲线下面积或发生剂量限制毒性反应,剂量爬坡方法就切换为传统的“3+3”药物临床试验设计。基于模型辅助设计和基于模型设计的临床试验方案具有相似的准确性,但更为简单易用。这类方法的特点是剂量增减的规则类似于传统的“3+3”药物临床试验设计,但是规则的制定则基于统计模型的估计。因此,其相较于传统的“3+3”药物临床试验设计具有更好的统计特性和适用性。
2 无缝设计的临床试验方案
近年来,新型药物研发面临着加速推进临床试验研究进度的需求,以在较短时间内得到相对确切同时满足统计学要求的研究结论,满足患者迫切的临床需求。然而,既往的确证性的Ⅲ期临床试验方案大多数须提供生存获益的疗效数据,研究周期较长,造成部分疗效好的药物或治疗方法无法早期用于患者而使其获益。在此背景下,“无缝设计”逐步取代传统的三阶段药物研发模式,成为加快获得研究结论的重要路径。例如,2015年上市的奥希替尼研发团队通过无缝Ⅰ/Ⅱ期设计,在Ⅰ期剂量爬坡观察安全性的同时观察治疗有效性(Ⅱ期),同时开展人群分层防止无效人群对结果的稀释,最后在2年时间内基于411例晚期非小细胞肺癌患者的2项单臂研究结果数据获得美国FDA的批准,节约了研发的周期。此外,在应用无缝Ⅱ/Ⅲ期设计消除Ⅱ期和Ⅲ期之间的空白期时,既可采用“推断无缝设计”在主要分析中纳入Ⅱ期试验受试者,也可以采用“操作无缝设计”,将Ⅱ期试验的受试者排除在主要分析之外。“推断无缝设计”须根据适应性的性质和假设检验策略作出相应的调整,而“操作无缝设计”则无须对Ⅰ类错误的控制进行多重性调整。近年来,新药研发模式也发生了巨大变革,使用扩展队列早期临床无缝试验设计,即通过动态处理数据、队列扩展等方法,将传统的分期试验(Ⅰ/Ⅱ/Ⅲ/Ⅳ期)压缩成单一的连续试验,以实现传统多个分期试验所需实现的目标,从而有助于提高研发效率、降低试验成本、缩短临床开发总时间。应强调的是,无缝设计并非只有优点,相对于传统分期设计,无缝设计方案无论是对研发还是对监管都提出了更为严格的要求,同时因试验快速推进带来的受试者伦理風险控制也须得到充分的考量。为更好地控制含有多个扩展队列的无缝试验的风险,增加扩展队列试验的安全性,美国FDA指南对申办方提出了4项建议:①监测和报告研究过程安全问题;②建立独立的安全评估委员会或独立的数据监测委员会;③研发团队与伦理审查委员会保持一致的沟通;④定期更新知情同意书。
3 篮式设计的临床试验方案
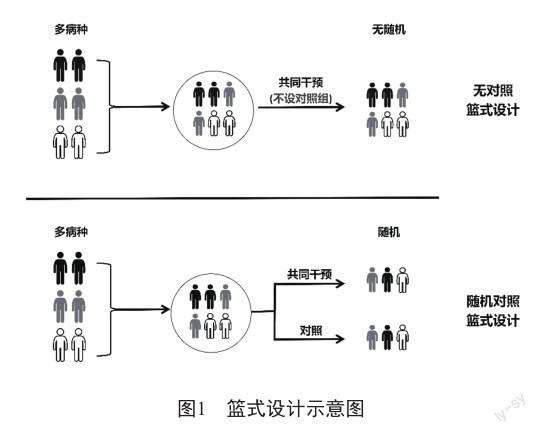
近年来,随着分子生物学技术的飞速发展,人类对疾病表型的认识不断深入,因此对疾病的治疗也逐渐步入基于分子表型的精准诊疗时代。精准医学不断发展,人类对疾病的认知则逐步精细化,例如过去根据发病部位和形态病理确定患者罹患某一种肿瘤,目前可被分割为数十种肿瘤;而过去不同种类的肿瘤,目前根据其分子生物学特征被划分为同一类肿瘤。传统的临床试验设计是一个临床试验方案仅在单一疾病人群中进行一两个药物或方案的试验。但大量涌现的疾病亚群存在研发速度上的局限性,尤其对于那些亚群患者数量较少的疾病就显得尤为滞后,须大量筛选患者和智能试验设计才能有效利用资源。在此背景下,篮式设计方案应运而生。
篮式设计旨在评估一种药物治疗具有同一种生物学特征的不同疾病类型的临床效果,每一个子方案都针对一种或多种传统疾病类型。如图1所示,篮式设计包括“无对照的篮式研究”和“同期随机对照的篮式研究”2种类型。篮式设计类似于中医学的“异病同治”理念,即用相同的治疗方案对发病机理相同的不同疾病进行统一治疗。须说明的是,这里所提及的“异病”是基于传统的按照发病部位和形态病理所作的诊断,篮式设计只是形象说法,其背后的实质是精准医学理念的临床实践。篮式设计的一个重要优势是“共享”,通过“异病同治”理念提高效率,加快药物开发速度。若按照传统临床试验方案,每个疾病亚型都要进行一次独立筛选,重复筛选将延缓患者入组的速度,患者甚至可能因为疾病的进展而失去了进入试验的机会。另外,篮式设计将搭建一个独立的框架或平台,其信息化系统、数据监管、毒性评估、疗效判定、试验决策的人员配置等均可做到中心化管理,除了高效外还可提高试验的质量,同质化的管理为后续基于较大样本进行高质量的转化研究提供了强有力的支撑。
4 伞式设计的临床试验方案
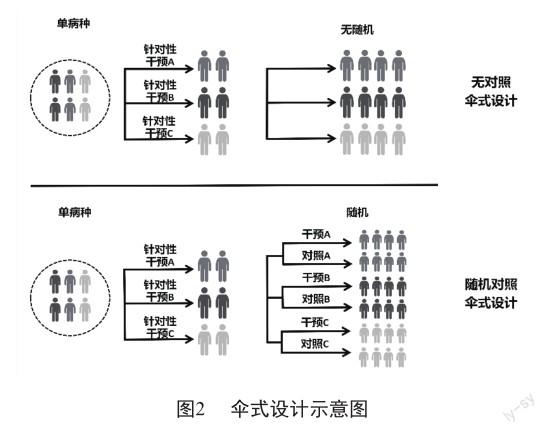
与篮式设计的临床试验方案类似,伞式设计也是在分子生物学技术的飞速发展,人类对疾病表型的认识不断深入的背景下出现的一种新型临床试验设计方案。伞式设计旨在评估多种药物针对同一种疾病但不同生物标记物类型患者的治疗效果。如图2所示,伞式设计包括“无对照伞式研究”和“随机对照伞式研究”2种类型。与篮式试验类似于“异病同治”不同,伞式设计是“同病异治”的方案设计。同样,这里的“同病”也是基于传统按照发病部位和形态病理所作的诊断,伞式设计只是形象说法,其背后的实质也是精准医学理念的临床实践。
伞式设计的临床试验方案也同样具有篮式设计的优点,包括通过“共享理念”提高研究效率,加快药物研发进度。同时,伞式设计也同样会搭建一个独立的框架或平台,其信息化系统、数据监管、疗效判定、试验决策的人员配置等均可做到中心化管理。此外,伞式设计还具有受试者共享的优势。临床研究受试者是临床试验最重要的资源,在随机对照临床试验设计的子方案采用多个子方案共享一个对照组毫无疑问可以节约受试者资源,这对于罕见疾病的患者群体尤为重要。目前,依据治疗反应而自适应性调整的随机临床试验不断受到关注,其在拥有多个子方案的伞式设计中会享有更大的游刃度。与篮式设计类似,伞式设计中同质化的管理也为后续基于较大样本进行高质量的转化研究提供了强有力的支撑。
5 主方案设计的临床试验方案
主方案设计是对传统篮式和伞式设计的进一步拓展,主要包括2个方法:①把篮式和伞式试验融合在一起的一体化设计;②从传统设计的单纯研究框架搭建转化成研究平台的搭建,即所谓的平台试验(platform trial),在这个平台上可添加新的药物和停止无效药物,没有固定的截止时间,具有永久性和动态性2大特点。主方案设计一般采用贝叶斯等自适应决策规则确定是否和何时停止具有低成功概率或有副作用的治疗方案,以及确定是否或何时将未来成功概率高的治疗方案推进研究。对于已有前期研究结果并具备条件的新药物或治疗方案可新纳入平台;对于已取得成功并获批适应证的药物和方案将调整成为平台相应子研究对照组。
主方案设计作为临床试验设计的重要创新,也存在着一些难点应在实践中考量。①相比于传统的临床试验,主方案设计所纳入的药物或方案多,涉及的药品研发企业也多,因此如何把这些不同企业的药品和资源进行合理整合是一个难题。②主方案设计所涉及的研究药物、方案,以及在整体研究中所入组患者数目和患者的异质性均会增加,给统计设计和分析也带来了挑战。如篮式设计试验中,虽然入组的是具有同一种生物学特征的人群,但按发病部位和形态病理的传统疾病分类也会对疗效产生影响,不合理的对照组参照可能因为疾病的混杂出现统计学效力的稀释和假高。平台试验的动态性自适应设计和决策的统计学方法目前还不够完善,尤其期中分析样本量较低,可能会导致错误结论。③不同的药物的毒效应谱不一样,多数药物的纳入会使得研究的毒性管理以及须根据毒性来评估进行方案决策的团队尤为重要,必须在临床试验前根据前期的研究数据慎重考虑,建议尽可能全地纳入相关学科的专家,并适时进行调整。
6 富集设计的临床试验方案
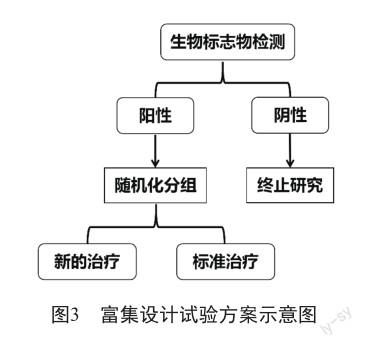
在精准医学时代,基于个体水平的分子表型测定变得切实可行,越来越多的临床试验选择以生物标志物为驱动,开展寻找试验药物的最佳获益人群与个性化管理方案。通过有效的生物标志物精准筛选潜在获益人群,有助于提高临床试验的成功率,同时还能避免将获益可能性小的患者人群暴露于不必要的安全性风险中[3]。在具备合理设计与充足资源保障的前提下,生物标志物驱动的临床研究可高效地为患者个性化治疗提供证据(图3)。
富集设计临床试验方案仅针对生物标志物阳性人群进行随机化分组后,给与干预治疗、评估干预方案的疗效和安全性。“富集”是指在临床试验中根据受试者的某些特征(如人口学、病理生理学、组织学、基因组学等)精准定义药物临床试验潜在的获益最大化目标人群。广义上,富集策略主要是指随机对照临床试验中用于选择最有可能获益的受试者的方法,但也可扩展到使用外部对照的单臂试验。常见的以生物标志物驱动的富集策略包括预后型富集、预测型富集、复合型富集和适应性富集等,研究者可根据临床试验关注的主要问题选择合适策略。
①预后型富集是通过对预后生物标志物的识别,入选更容易出现预后结局或疾病进展的人群,该策略可增加试验的绝对效应。预后因素的判断只须通过看无治疗或在对照组治疗下,生物标志物阳性与阴性患者发生终点事件的风险是否有差别。②预测型富集则是根据疾病特征选择对试验药物最可能有应答的受试者以提高试验效率,该策略既可增加试验的绝对效应,也可增加相对效应,能以较小的样本量获得较高的检验效能。预测因素的判断须通过比较生物标志物阳性人群中的药物疗效与生物标志物阴性人群中的药物疗效之间的差异。值得注意的是,预后型与预测型并不是绝对的,某研究中是预后型的因素在其他研究中也可以作为预测型出现。③复合型富集是指同时使用多个生物标志物(例如综合评分的形式)以减少受试者异质性来提高试验效率的策略。④适应性富集则是按照预先设定的计划,在保证试验合理性和完整性前提下,根据试验期中分析结果对目标人群进行适应性调整,例如改变入组标准或仅纳入一个生物标志物亚组的受试者等。
富集设计针对生物标志物阳性人群可以高效地提供获益风险证据,一般所需的样本量也较小。然而,值得注意的是,采用富集设计还应考虑以下几个关键问题:①生物标志物检测的灵敏度和特异度,应该对选择高风险或对试验药物有应答的受试者具有较高的灵敏度,同时对鉴别低风险或对试验药物无应答的受试者有较高的特异度。当生物标志物检测准确性不够时,假阴性会导致入组效率较低,假阳性又会稀释研究疗效。②试验结果在生物学上是否具有可解释性或可重现性,以及临床实践中的可推广性。③研究结果的适用性与外推性是富集策略的主要问题,当存在生物标志物检测阈值不确定时,可以考虑入组更宽泛的阳性人群,然后基于不同检测阈值进一步做分层随机。④富集设计不能进一步探索生物标志物的预测性,为了克服这个局限性,实际上有些试验采取了更灵活的设计形式,比如同时设计一个阴性人群的独立队列,在不影响主要队列分析的情况下,提供进一步探索生物标志物的预后和预测性以及药物在阴性人群中的获益风险比的可能性。
7 生物标志物分层设计的临床试验方案
生物标志物分层临床试验设计也是在精准医学时代基于生物标志物进行分层后再开展随机化分组,实际是针对全人群的随机化试验方案(图4)。同样,生物标记物分层设计通过有效的生物标志物精准筛选潜在获益人群。在具备合理设计与充足资源保障的前提下,生物标志物分层设计临床研究可高效地为患者个性化治疗提供证据。须注意的是,当存在多个生物标志物时,一项试验也可以组合不同的基本设计,比如针对一个生物标志物进行富集,针对另一个生物标志物进行所有人群随机并分层。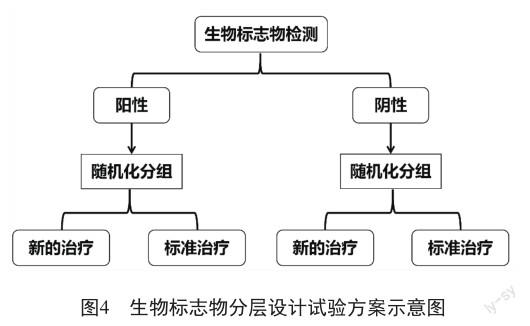
生物标志物分层设计的临床试验方案中,根据功能特点不同,与药物研发相关的生物标志物可分为多个类型,包括诊断性、预后性、预测性、药效学、安全性和监测性生物标志物。其中,预后性生物标志物常常在临床试验中被用作分层因素,可以区分未接受治疗干预下诊断相同但自然进程不同的患者人群。在对照研究中利用预后性生物标志物可以降低受试者的异质性和混杂因素对试验结果的干扰,减少组间偏倚,提高结果可靠性。对于前期已有充分的基础研究数据支持但未经临床验证的预测性生物标志物,通常不建议仅在生物标志物阳性患者中开展新药试验,而建议将生物标志物作为分层因素同时纳入阳性与阴性患者人群进行研究。当生物标志物检测不成熟时,入组全人群可以支持针对不同生物标志物检测方法和阈值的探索性分析。但是,当生物标志物阳性率较低或阴性人群获益非常有限时,试验效率也会低,通常需要更大的样本量和更大的全人群检测成本。
生物标志物分层设计经常会涉及主要研究终点是阳性亚组还是整体人群的多重性问题[4]。当设计以阳性亚组作为主要研究终点之一或关键次要终点,并且设计了足够的样本量,研究结果有足够把握度证明生物标志物阳性亚组人群能够从新药治疗中显著获益,则该研究结果可基于生物标志物支持批准试验药物在阳性亚组人群中的适应证。值得注意的是,设计多个主要研究终点时,还应考虑合适的策略保证整体Ⅰ类错误a不增加,可以采取平行策略对Ⅰ类错误进行拆分、回收或采取固定次序序贯检验策略。临床研究实践发现,常见的先检验生物标志物阳性人群再检验生物标志物阴性人群或全人群的序贯策略不一定是最优选择,生物标志物的阳性率、检测准确性以及公司投资风险的判断等都可能是统计分析决策的考量点之一。当采用固定次序检验策略时,第一层检验的效度对整体研究是否成功至关重要。如果生物标志物检测阳性率低,那就不适合作为第一层进行设计,因为研究检验效能既受疗效影响,也受事件数/样本量的影响,所以相同随访时长下生物标志物阳性亚组不一定有更高的检验效能。
参考文献
[1] 王瑞平, 肇晖, 李斌. 随机对照临床试验设计要点和规范[J]. 上海医药, 2022, 43(7): 72-77.
[2] 王瑞平, 李斌. 临床研究理论规范和实践[M]. 上海: 上海科学技术出版社, 2023.
[3] 国家药品监督管理局药品審评中心. 药物临床试验富集策略与设计指导原则(试行)[EB/ OL]. (2020-12-31)[2023-03-01]. https://www.cde.org.cn/zdyz/domesticinfopage?zdyzI dCODE=dae0b296a6ca491b977434b1ad220936.
[4] 国家药品监督管理局药品审评中心. 药物临床试验多重性问题指导原则(试行)[EB/OL]. (2020-12-31)[2023-03-01]. https://www.cde.org.cn/zdyz/domesticinfopage?zdyzIdCODE=7e708c2bed01197927a864f3c3e8c972.
