糖尿病因素削弱右美托咪定对肾缺血再灌注后心肌损伤的保护作用
2023-07-06伊合山艾尼瓦尔张静静祖力亚尔吾斯曼
耿 强, 伊合山·艾尼瓦尔, 张静静, 祖力亚尔·吾斯曼, 南 乐, 张 冰
(新疆医科大学1第三临床医学院, 2附属肿瘤医院麻醉科, 乌鲁木齐 830011)
糖尿病(Diabetes mellitus,DM)是由于氧化应激、胰岛素抵抗引起的一系列代谢综合征[1],其患病率呈现快速增长的态势[2]。糖尿病靶器官损害是导致糖尿病患病人群死亡的重要诱因。研究表明,罹患糖尿病后心肌损伤的敏感性增高[3]。
本课题组前期研究发现[4],肾缺血再灌注损伤(Renalischemia-reperfusioninjury,RI/RI)能够激活核因子-κB(Nuclear factor-κB,NF-κB)诱发心肌损害,而右美托咪定预处理(Dexmedetomidinepreconditioning,DP)能够为其提供有效的心肌保护,炎症机制是其中心环节[5],但在2型糖尿病背景下右美托咪定对心肌保护作用的有效性以及是否涉及炎症机制目前尚未明确。本研究拟通过制备2型糖尿病大鼠肾缺血再灌注(RIR)后心肌损伤模型,并与非2型糖尿病大鼠模型对比,探究糖尿病因素削弱右美托咪定对RIR后心肌保护的炎症机制。
1 材料与方法
1.1 动物模型制备及分组SPF级健康雄性Wistar大鼠,12周龄,体重200~300 g,由新疆医科大学动物实验中心提供,本实验经新疆医科大学实验动物伦理委员会审查批准实施(审批号:IACUC-20220306-05)。大鼠均饲养在屏障环境中,自由摄食饮水。
取Wistar大鼠30只,适应性饲养后给予45%高糖高脂饲料喂养4 w。禁食12 h,腹腔注射1%链脲佐菌素(STZ)30 mg/kg,72 h后测量随机血糖。若出现随机血糖≥16.7 mmol/L以及多饮、多尿、体重减少症状则认为2型糖尿病模型建模成功。
在构建大鼠2型糖尿病模型期间有6只大鼠死亡,故24只大鼠建模成功,随机分为模型+盐水组(DM-C组)、模型+肾缺血再灌注组(DM-IR组)和模型+右美托咪定预处理组(DM-DP组)。另取24只非2型糖尿病大鼠作为对照并随机分为正常+盐水组(NDM-C组)、正常+肾缺血再灌注组(NDM-IR组)和正常+右美托咪定预处理组(NDM-DP组)。(1)模型+盐水组和正常+盐水组大鼠只开腹,尾静脉泵注等量生理盐水;(2)肾缺血再灌注处理:术前禁食8 h,不禁水,称重。腹腔注射10%水合氯醛350 mg/kg麻醉后固定,沿腹白线开腹约5 cm,暴露腹腔。分离并切除右肾,再暴露左肾及肾蒂,分离左肾动脉,以动脉夹夹闭左肾动脉,可见左肾颜色由鲜红变为暗红,则肾缺血成功,缺血45 min后恢复动脉血供,若肾脏颜色逐渐由暗红变成鲜红色表明肾缺血再灌注建模成功,尾静脉持续泵注等量生理盐水。操作完成后关闭腹腔,持续体外加温并计时。(3)右美托咪定预处理:开放尾静脉通路后,按体表面积法,以2 μg·kg-1·h-1的速率于缺血前30 min起泵注至再灌注结束后4 h。
1.2 主要药品及试剂盐酸右美托咪定注射液(批号:21051531,扬子江药业集团有限公司,中国),链脲佐菌素STZ(CAS号:18883-66-4,Sigma,美国),45%高脂饲料(45%脂肪、0.2%胆固醇),柠檬酸钠缓冲液(批号:C1013,Solarbio,中国),肌钙蛋白I(cTnI)(华美,中国),肿瘤坏死因子-α(TNF-α)(联科,中国),白介素-17a(IL-17a)(联科,中国),白介素-10(IL-10)(联科,中国),核因子-κB(NF-κB p65)(博奥森,中国),p-NF-κB p65 (Ser536) (博奥森,中国),β-actin(义翘神州,中国)。
1.3 标本采集再灌注结束后,将大鼠背部垫高,开腹,游离腹主动脉,使用静脉采血针留取血标本。同时,利用失血性休克法处死大鼠后,开胸分离心脏,沿主动脉根部剪下心脏,置入4℃PBS中清洗干净,留取左心室,分装如冻存管后置入液氮中保存。每组中随机选取3例进行后续检测。
1.4 ELISA法检测血清中cTnI、IL-17a、IL-10水平将血标本混匀、静置30 min,离心(2 000 r/min,15 min,4 ℃)后取上清,采用ELISA法测定血清中cTnI、IL-17a、IL-10水平。
1.5 TUNEL染色观察心肌组织凋亡水平并计算凋亡率使用TUNEL凋亡检测试剂盒-POD进行TUNEL分析以评估细胞凋亡。心肌组织使用甲醛溶液固定并包埋在石蜡中,将组织切成5 μm切片进行TUNEL染色。通过在生物显微镜下观察并以盲法计算和分析每个高倍视野的TUNEL阳性细胞数,计算凋亡率。
1.6 Western blot法检测心肌组织中NF-κBp65的表达及磷酸化水平心肌组织样本经液氮研磨后,加入400 μL RIPA裂解液,充分混匀。4℃静置60 min后,4℃离心15 min并收集上清,采用BCA法对所提蛋白定量,考马斯亮蓝染色。使用5×SDS-PAGE电泳,转入PVDF膜上,加入5%脱脂奶粉的封闭液,封闭转印膜1 h。加入TBST稀释一抗(稀释度1∶400,博奥森公司),4℃孵育过夜。TBST洗膜3次,加入山羊抗兔IgGHRP(稀释度1∶5 000,Abcam公司),室温孵育1 h。TBST洗膜3次。将显色液A液和B液混匀,取2 mL加至膜上。使用Chemiscope 3000检测、拍照。通过ChemiAnalysis软件计算得到条带灰度值,利用目的蛋白与内参蛋白的比例计算得到蛋白的相对表达量。
2 结果
2.1 各组大鼠血清cTnI水平比较与NDM-C组比较,NDM-IR组cTnI水平升高(P<0.05);与NDM-IR组比较,NDM-DP组cTnI水平降低(P<0.05);与DM-C组比较,DM-IR组和DM-DP组cTnI水平均升高(P<0.05);与DM-IR组比较,DM-DP组cTnI水平降低(P<0.05);与NDM-DP组比较,DM-DP组cTnI水平升高(P<0.05)。见表1。
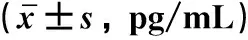
表1 各组大鼠血清cTnI水平比较
2.2 各组大鼠血清IL-17a、IL-10水平比较与NDM-C组比较,NDM-IR组IL-17a水平升高、IL-10水平降低(P<0.05);与NDM-IR组比较,NDM-DP组IL-17a水平降低、IL-10水平升高(P<0.05);与DM-C组比较,DM-IR组和DM-DP组IL-17a水平均升高、IL-10水平均降低(P<0.05);与DM-IR组比较,DM-DP组IL-17a水平降低、IL-10水平升高(P<0.05);与NDM-DP组比较,DM-DP组IL-17a水平升高、IL-10水平降低(P<0.05)。见图1。
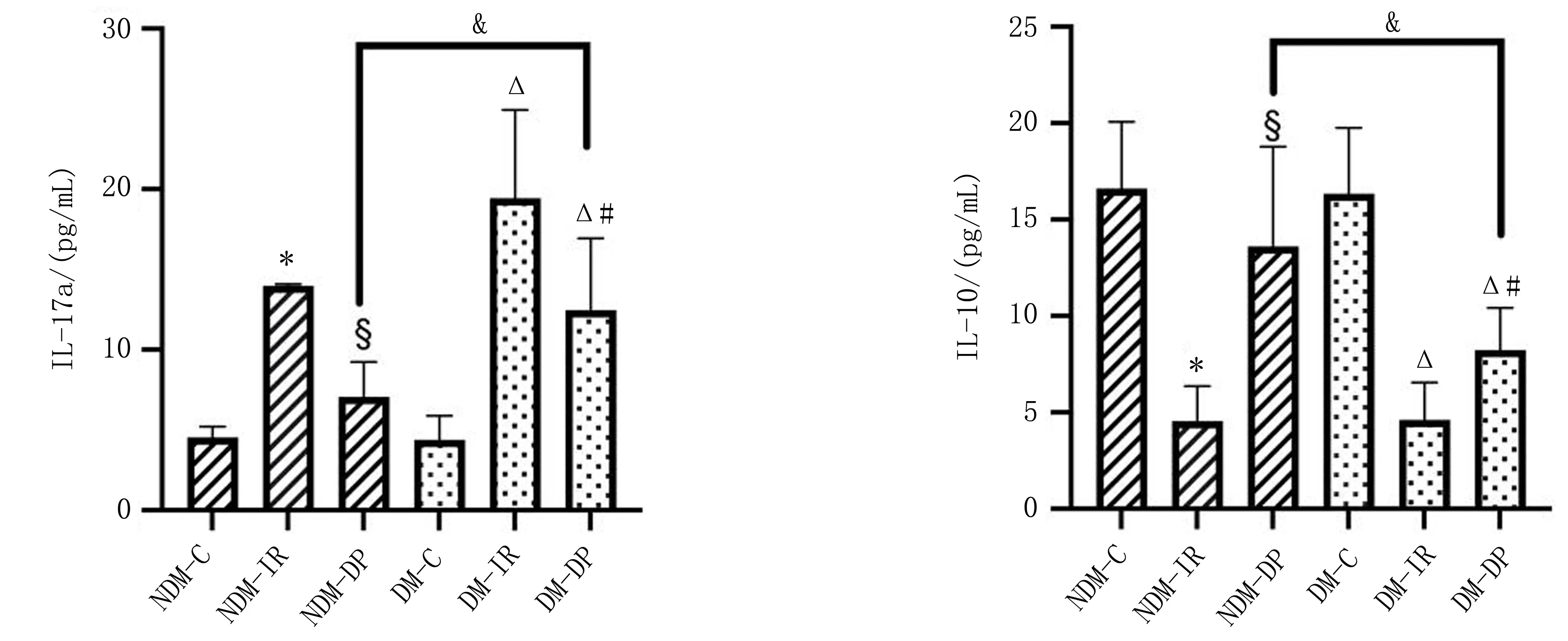
注:与NDM-C组相比, *P<0.05; 与NDM-DP组相比, &P<0.05; 与NDM-IR组相比, §P<0.05; 与DM-C组相比, △P<0.05; 与DM-IR组相比, #P<0.05。图1 各组大鼠血清IL-17a、IL-10水平比较
2.3 各组大鼠心肌组织TUNEL染色和凋亡率比较与NDM-C组比较,NDM-IR组凋亡率升高(P<0.05);与NDM-IR组比较,NDM-DP组凋亡率降低(P<0.05);与DM-C组比较,DM-IR和DM-DP组凋亡率升高(P<0.05);与DM-IR组比较,DM-DP组凋亡率降低(P<0.05);与NDM-DP组比较,DM-DP组凋亡率升高(P<0.05),见表2。TUNEL染色结果见图2。
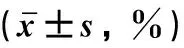
表2 各组大鼠心肌组织凋亡率比较
2.4 各组大鼠心肌组织NF-κBp65蛋白表达及磷酸化水平的比较与NDM-C组比较,NDM-IR组NF-κB p65表达增高(P<0.05);与NDM-IR组比较,NDM-DP组NF-κB p65表达降低(P<0.05);与DM-C组比较,DM-IR组和DM-DP组p-NF-κB p65升高(P<0.05);与DM-IR组比较,DM-DP组p-NF-κB p65降低(P<0.05);与NDM-DP组比较,DM-DP组NF-κB p65、p-NF-κB p65增高(P<0.05);NDM各组间p-NF-κB p65水平和DM各组间NF-κB p65表达差异无统计学意义(P>0.05)。见图3。
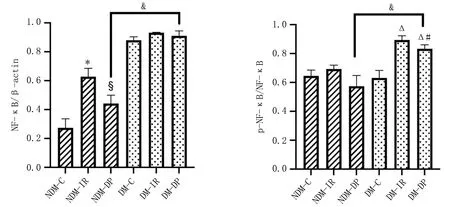
注:与NDM-C组相比, *P<0.05; 与NDM-DP组相比, &P<0.05; 与NDM-IR组相比, §P<0.05; 与DM-C组相比, △P<0.05; 与DM-IR组相比, #P<0.05。图3 各组大鼠心肌组织NF-κBp65、p-NF-κBp65蛋白的表达比较
3 讨论
本实验通过选用2型糖尿病大鼠和非2型糖尿病大鼠分别建立RIR模型,研究2型糖尿病因素削弱右美托咪定预处理对大鼠RIR后心肌损伤的保护效果的炎症机制。研究发现,右美托咪定一定程度上能够降低2型糖尿病大鼠RIR后心肌组织凋亡率,降低心肌损伤标志物cTnI的活性,通过干预NF-κB调节炎症因子IL-17a与IL-10的含量,降低心肌凋亡率,但与右美托咪定干预非2型糖尿病大鼠RIR后心肌组织损伤情况对比,发现2型糖尿病是削弱右美托咪定预处理RIR后心肌损伤保护的关键因素。
cTnI的检测已被证实对早期发现心肌损伤的特异性和敏感性方面具有独特的优越性,是判断心肌损伤的基石[6-7]。IL-17a、IL-10是参与体内炎性反应的重要炎性因子。本研究结果显示,在非2型糖尿病大鼠中,RIR发生后IL-17a水平升高,抗炎因子IL-10水平降低,血清cTnI水平升高,心肌凋亡率明显升高,表明RIR后会发生心肌损伤,对其实施右美托咪定预处理后,血清IL-17a水平降低,IL-10水平升高,cTnI水平降低,心肌凋亡率显著降低,均提示右美托咪定预处理能够有效缓解RIR带来的心肌损伤,这与本课题组前期研究一致[8]。在2型糖尿病大鼠中,RIR发生后,心肌损伤的结局与非2型糖尿病大鼠RIR后类似,表明右美托咪定预处理依然能够发挥心肌保护作用,然而对2型糖尿病大鼠肾缺血再灌注即便实施右美托咪定预处理后,IL-10水平和凋亡率仍未恢复至2型糖尿病对照组水平。提示2型糖尿病因素阻碍了右美托咪定预处理对肾缺血再灌注后的心肌保护效应。
炎症介质在多种器官损伤[9-11]的病理生理机制中占重要地位。炎症介质能够在器官和血清中积累,触发心肌凋亡[12]。本研究中,非2型糖尿病大鼠肾缺血再灌注组NF-κB p65表达较非2型糖尿病大鼠对照组明显增加,而非2型糖尿病大鼠各组间p-NF-κB p65水平均无统计学差异,在2型糖尿病大鼠各组间NF-κB p65水平差异无统计学意义,反映了2型糖尿病因素使得机体始终处于炎症状态,NF-κB p65的表达阈值上调。同时,即便对2型糖尿病RIR大鼠实施DP,无论是NF-κB p65还是p-NF-κB p65均较非2型糖尿病RIR大鼠实施DP升高,提示2型糖尿病因素削弱了DP的保护作用。Itoh等[13]学者发现,IL-17a激活NF-κB,使其磷酸化,并刺激促炎因子如IL-6、TNF-α和GM-CSF诱导炎症,进而参与糖尿病心肌的损伤。研究发现NF-κB激活后,诱导产生大量促炎细胞因子,造成器官损害[14]。说明NF-κB和炎症因子的串扰加剧了靶器官损害。
本研究选用高糖高脂饮食联合STZ注射的方法构建2型糖尿病模型。2型糖尿病往往伴随严重的胰岛素抵抗,疾病发生后降低了胰岛素的心肌保护作用[15],造成“心肌敏化”,当遭受远隔器官缺血再灌注后,损伤往往更为严重,其机制可能涉及糖代谢关键通路[16],激活NF-κB,诱发机体的慢性炎症状态,这可能也是阻碍右美托咪定预处理发挥抗炎作用的原因所在。
综上,炎症机制参与了糖尿病因素削弱右美托咪定预处理对RIR后心肌的保护作用。下一步,本研究将继续探寻炎症的关键信号通路,更完善地发挥右美托咪定预处理的效能,为临床转化提供科学依据。
