气相钝化对FeS自燃过程特征阶段的影响
2023-06-29高建村贾少康
高建村, 贾少康, 许 芹
(1.北京石油化工学院 安全工程学院, 北京 102617; 2.北京市安全生产工程技术研究院, 北京 102617)
0 引 言
随着石油化工行业的发展,我国原油需求量不断升高[1],高含硫原油中的硫组分(单质S、H2S和有机硫)会对炼油设备、储存装置和运输管道造成严重的腐蚀破坏,形成的硫腐蚀产物主要有FeS、FeS2、Fe3S4等。其中,较为常见的硫腐蚀产物是FeS,具有室温自燃活性,一旦接触空气即自燃产生热量,极易引发火灾及爆炸事故,严重威胁石油化工领域安全生产[2-4]。
为了有效防治硫化亚铁自燃问题,当前主要采用隔离、清洗及钝化等防治技术[5-7],隔离与清洗防治技术存在费用高、清理废物需多次处理、液相清洗效率不高等缺点。Walker等[8-9]率先提出使用氮氧混合气体钝化活性FeS的方法。该方法能够清洁、有效地将自燃活性FeS转变为较为稳定的铁氧化合物,降低FeS自燃风险。宋红荣等[10]相继通过水蒸气与钝化气体混合形成的低氧混合气进行钝化。赵雪娥[11]首次提出低流速状态下的空气进行钝化的方法,两者都能有效降低FeS氧化反应放热速率,能够起到一定的钝化效果。Asaki等[12-13]认为当采用富氧气体钝化FeS,生成铁氧化合物及二氧化硫,但FeS存在自燃安全隐患,低氧浓度下钝化FeS相对稳定安全,钝化产物为铁氧化合物与单质硫,能够有效避免FeS自燃。代濠源等[14]通过配比不同氮氧混合气进行硫化亚铁钝化,当钝化气氛中氧浓度降低至1.25%时,整个钝化过程放热较小,更加安全,但钝化时间相对较长。以上研究表明,低氧混合气体能够将活性FeS钝化为较为稳定的铁氧化合物,但FeS自燃过程及气相钝化抑制机理尚未清晰,制约FeS自燃气相钝化防治技术的发展。
鉴于以上问题,笔者通过液相合成法制备室温下具有自燃活性的FeS,采用气相钝化装置对活性FeS进行钝化处理,借助同步热分析仪,研究气相钝化对活性FeS自燃过程特征阶段的影响,从自燃反应动力学角度揭示气相钝化对FeS自燃的抑制机理,对硫腐蚀产物气相钝化防治技术的发展具有重要意义。
1 实 验
1.1 设备
气相钝化装置主要由反应釜主体、进出气管路、流量计和气体浓度监测等构成,采用计算机连接设置钝化参数。X射线衍射仪由德国布鲁克AXS有限公司生产,型号为D8,衍射角度范围0~130°。SDT-Q600同步热分析仪的水平双臂的测定范围为0.001~200 mg,量热精度±2%,重量灵敏度≤0.1 μg,温度-1 500 ℃,对样品的测量准确度高于0.1%,升温速率范围0.1~100 ℃/min。
1.2 实验过程
由图1可见,通过液相合成法,以硫酸亚铁铵与硫化钠为原料,通过合成、洗涤和真空干燥等过程,得到室温下能够自燃的活性FeS,并密封保存[15-16]。在手套箱内将活性FeS样品置于气相钝化装置密封反应釜内,安装反应釜并连接进气与出气管线,通过计算机设置钝化气体流量为10 mL/min,对活性FeS进行持续1 h的气相钝化,得到钝化FeS并密封保存。对于活性与钝化FeS,整个取样过程在手套箱内操作,避免样品接触空气。在手套箱内称取实验样品质量10 mg置于小坩埚内,将坩埚置于SDT-Q600同步热分析仪的样品池,设置程序升温参数,实验气氛为干空气,气体流量为50 mL/min,温度为室温至420 ℃,升温速率分别为4、6、8、10 ℃/min,得到活性与钝化两组FeS样品,在不同升温速率下由室温氧化升温至420 ℃下TG-DSC曲线。对活性FeS,钝化FeS与热重实验后的FeS进行X射线衍射分析,衍射角度设定10°~90°,扫描速度为2(°)/min。
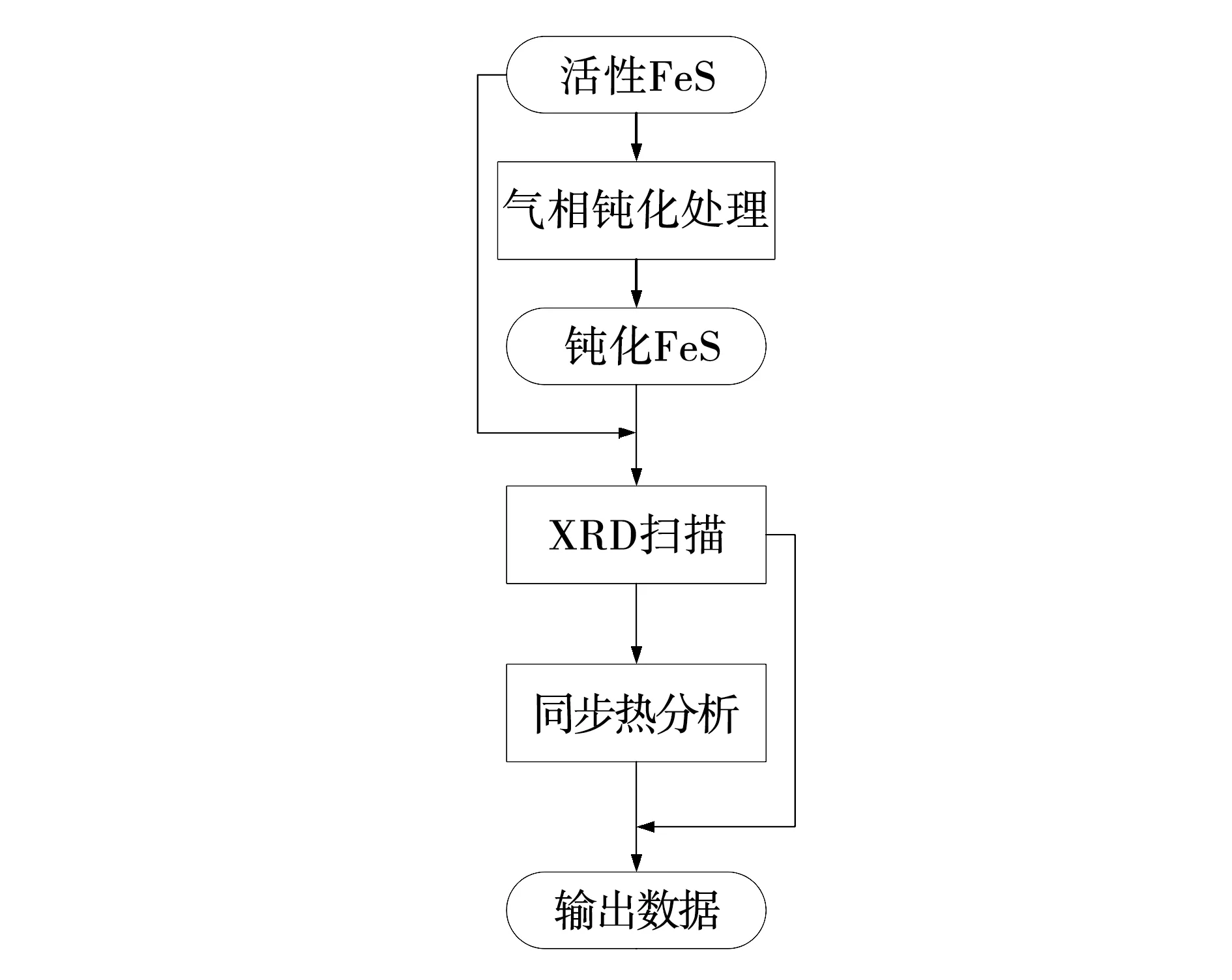
图1 实验流程Fig. 1 Experimental process
2 结果分析
2.1 气相钝化对活性FeS结构组分影响
FeS的XRD衍射图及标准谱图,如图2所示。活性FeS衍射峰不明显,在衍射角30°、43°、52°时出现少数稍显尖锐的衍射峰。主要是液相法制备的FeS样品结晶度较差,无法形成稳定的晶型结构,呈无定形态。对比标准谱图发现,气相钝化处理后的FeS在衍射角330°、44°时出现明显的衍射峰,钝化FeS由于与氧气反应生成了部分S与Fe2O3,但含量不高。经过热重实验后,出现更多的衍射峰,对比标准图谱发现,热重实验后样品中存在更多的Fe2O3。由于硫化亚铁经过热重实验后,与氧气充分反应,硫化亚铁进而转变为Fe2O3。

图2 FeS的XRD衍射图Fig. 2 XRD pattern of reactive FeS
2.2 FeS自燃过程的TG-DSC曲线
活性FeS与气相钝化FeS样品由室温升温至420 ℃自燃过程的TG-DSC曲线,如图3所示。升温速率为4 ℃/min,反应气体为干空气。结合TG-DSC曲线可以将活性FeS自燃过程分为四个阶段:自燃、吸氧增重、分解燃烧和燃尽。活性FeS在室温下开始自燃放热,出现第一个放热峰,TG曲线表现为失重的过程。随后进入吸氧增重阶段,出现第二个放热峰,持续至150 ℃左右,该阶段TG曲线表现为增重趋势。从150~250 ℃,FeS进入分解燃烧阶段,剧烈燃烧大幅失重的同时伴随着大量热量释放。随后进入燃尽阶段,质量在整个自燃过程末期基本保持不变,不继续放热。相较于活性FeS,气相钝化FeS的热流曲线在其第一阶段自燃的放热峰已经消失,室温下FeS不存在自燃现象,该阶段样品出现小幅失重。随后进入吸氧增重阶段,钝化FeS增重量较活性样品减小,放热量相对减小。150 ℃左右开始进入分解燃烧阶段,钝化FeS放出热量并失重,最终进入燃尽阶段。这说明气相钝化处理能够有效地抑制活性FeS室温下自燃的能力,显著降低了其自燃活性,减弱其吸氧增重阶段吸氧能力,延长分解燃烧过程,推移进入燃尽阶段的温度。

图3 升温速率4 ℃/min下FeS自燃过程的TG-DSC曲线Fig. 3 TG-DSC curves of FeS at a heating rate of 4 ℃/min
在不同升温速率下自燃过程的TG曲线,如图4所示。在自燃初期,活性FeS质量下降,出现第一个失重峰,其主要为样品室温下与氧气结合发生自燃,同时伴随着内部的水分蒸发及表面少量气体脱附。之后开始吸氧增重阶段,此阶段是FeS自燃过程的中期加速阶段,该阶段活性FeS质量开始逐步增加,活性FeS表面吸附活性位点吸附氧气,而此过程中氧化反应的耗氧量小于其吸附氧气量,总体表现为质量不断增加。在150 ℃左右吸氧增重达到最大值,随后快速分解燃烧,伴随着产物生成与气体的释放,呈现为快速失重状态,在220 ℃左右进入燃尽阶段,质量基本不变。钝化后的FeS氧化初期也出现了先失重后增重的现象,但其吸氧增重阶段的峰值与吸氧增重量明显低于活性样品。经过较为缓和的吸氧增重后开始燃烧分解,质量大幅降低,相较于活性FeS,钝化FeS进入燃尽阶段后移约30 ℃,整体反应进程相对减慢。升温速率影响着活性FeS的TG曲线,在室温升至50 ℃时,活性FeS的TG曲线受升温速率的影响并不明显,不同升温速率下的TG曲线图基本重合,但在分解燃烧阶段,TG曲线随着升温速率的增大呈向高温区迁移的现象。
不同升温速率下活性与钝化FeS自燃过程的DSC曲线,如图5和6所示。活性FeS在反应初期室温至50 ℃时,出现第一个明显放热峰,活性FeS的Fe具有未成对的d电子,d电子有较强的化学吸附性,与空气接触后,氧分子被FeS表面的活性中心吸附。多孔疏松的物质,与空气接触后发生吸附,在低温时发生氧化反应放出热量。随后进入吸氧增重阶段出现第二个放热峰,随着温度继续升高,FeS开始燃烧分解,在 150~250 ℃阶段放热量快速增加,呈现出更大的放热峰。相较于活性FeS,钝化FeS在室温至50 ℃范围内,室温下自燃的放热峰完全消失,放热初始温度移至180 ℃,分解燃烧放热最高峰的温度比活性FeS后移50 ℃,峰值高度也明显降低,整个自燃过程出现向高温段推迟的现象。由于经过气相钝化处理后,FeS表面形成了一层致密的氧化膜,降低了其与氧气初次结合进行反应的能力,无法室温下自燃。同时,随着升温速率升高,钝化FeS在放热最高处的峰值随着升温速率增加而增大。

图5 不同升温速率下活性FeS 自燃过程的DSC曲线Fig. 5 DSC curves of reactive FeS at different heating rates

图6 不同升温速率下钝化FeS 自燃过程的DSC曲线Fig. 6 DSC curves of passivated FeS at different heating rates
不同升温速率下活性与钝化FeS自燃过程的放热量占比曲线,如图7所示。两种样品的放热量占比曲线都呈现逐步增加后持平再迅速增加至不再放热的过程。活性FeS在室温至150 ℃放热速度明显高于钝化FeS,吸氧增重阶段结束,放热量占比约40%,也高于钝化FeS。说明钝化FeS在自燃过程初期,与氧气结合能力下降。分解燃烧阶段,两种样品放热速率、剧烈程度基本相似,但钝化FeS结束放热时间较钝化FeS延迟30 ℃左右,主要由于钝化FeS氧化阶段的诱导期延长,促使整个反应进程向高温段后移。随着升温速率升高,钝化FeS达到最大放热量的温度随之增加。
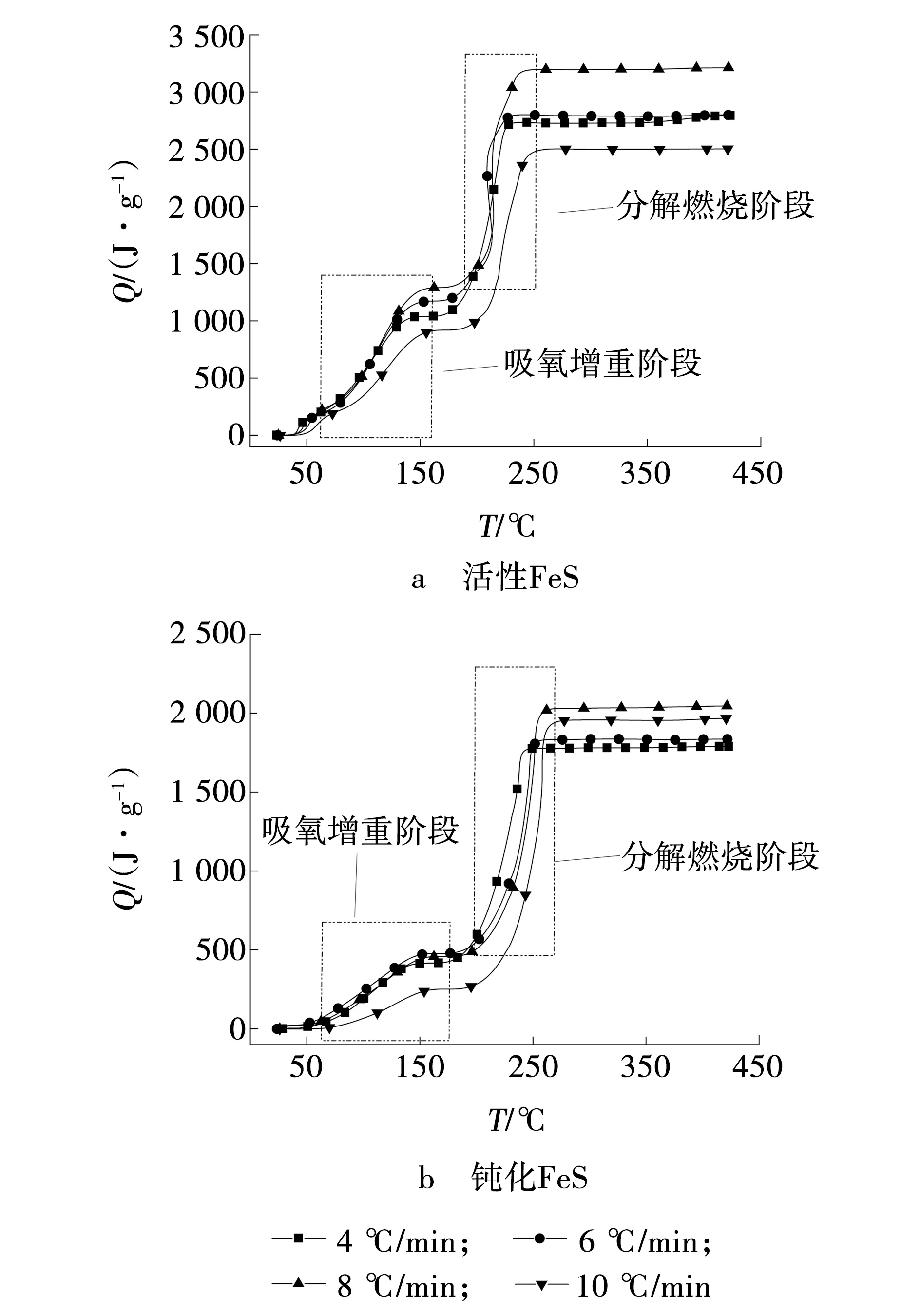
图7 活性与钝化FeS 自燃过程的放热量曲线Fig. 7 Heat released curves of reactive and passivated FeS
2.3 气相钝化时FeS室温自燃与吸氧增重
活性FeS室温下出现自燃现象,出现小幅失重与放出热量,该阶段是FeS自燃的初始过程,持续温度范围由室温开始至50 ℃左右。活性FeS与钝化FeS在室温自燃阶段失重量ws与放热量占比wf,如图8所示。活性样品失重量在4%~5%范围内,钝化FeS在该阶段也出现失重现象,失重量比活性样品小。钝化FeS在此阶段已经不自燃,其放热量基本为0,由于钝化后FeS活性降低,室温与氧气结合的能力降低,致使其基本不放出热量。
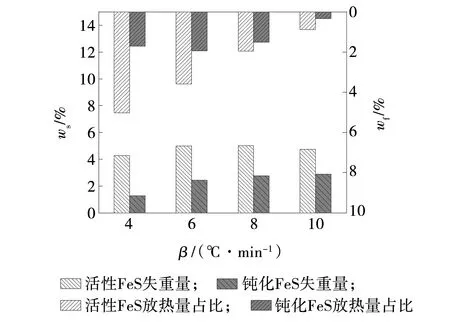
图8 FeS室温自燃阶段失重量及放热量占比Fig. 8 Mass loss and heat released proportion of FeS at spontaneous combustion stages at room temperature
活性FeS室温自燃后进入吸氧增重阶段。该阶段是FeS自燃过程的中期加速期,FeS吸附氧气与反应耗氧同时进行,主要表现为质量增加与持续放热[17-18]。活性FeS与钝化FeS在不同升温速率下吸氧增重持续温度范围与温度差值,如图9所示。
活性FeS吸氧增重阶段持续温度范围随着升温速率的增大而升高,说明升温速率增加能够延长活性FeS吸氧增重阶段。钝化样品吸氧持续温度范围显著降低,随着升温速率增加而降低。在10 ℃/min的升温速率下,钝化FeS的吸氧增重持续温度为71.64 ℃,相比活性FeS降低了35.57 ℃,说明气相钝化处理能够有效降低FeS自燃过程吸氧增重阶段持续时间。FeS钝化前后吸氧增重阶段温度范围差值随升温速率增加而增大,由于气相钝化处理后,钝化膜抑制了FeS吸附氧气能力,并且随着升温速率加快,热传递效益呈削弱趋势,因此,更不利于钝化后的FeS吸附氧气。
FeS吸氧增重阶段是吸附氧气与耗氧同时进行,吸附氧气量大于反应耗氧量,表现为质量增重与放出热量。钝化前后FeS样品在不同升温速率下吸氧增重量及放热量占比,如图10所示。
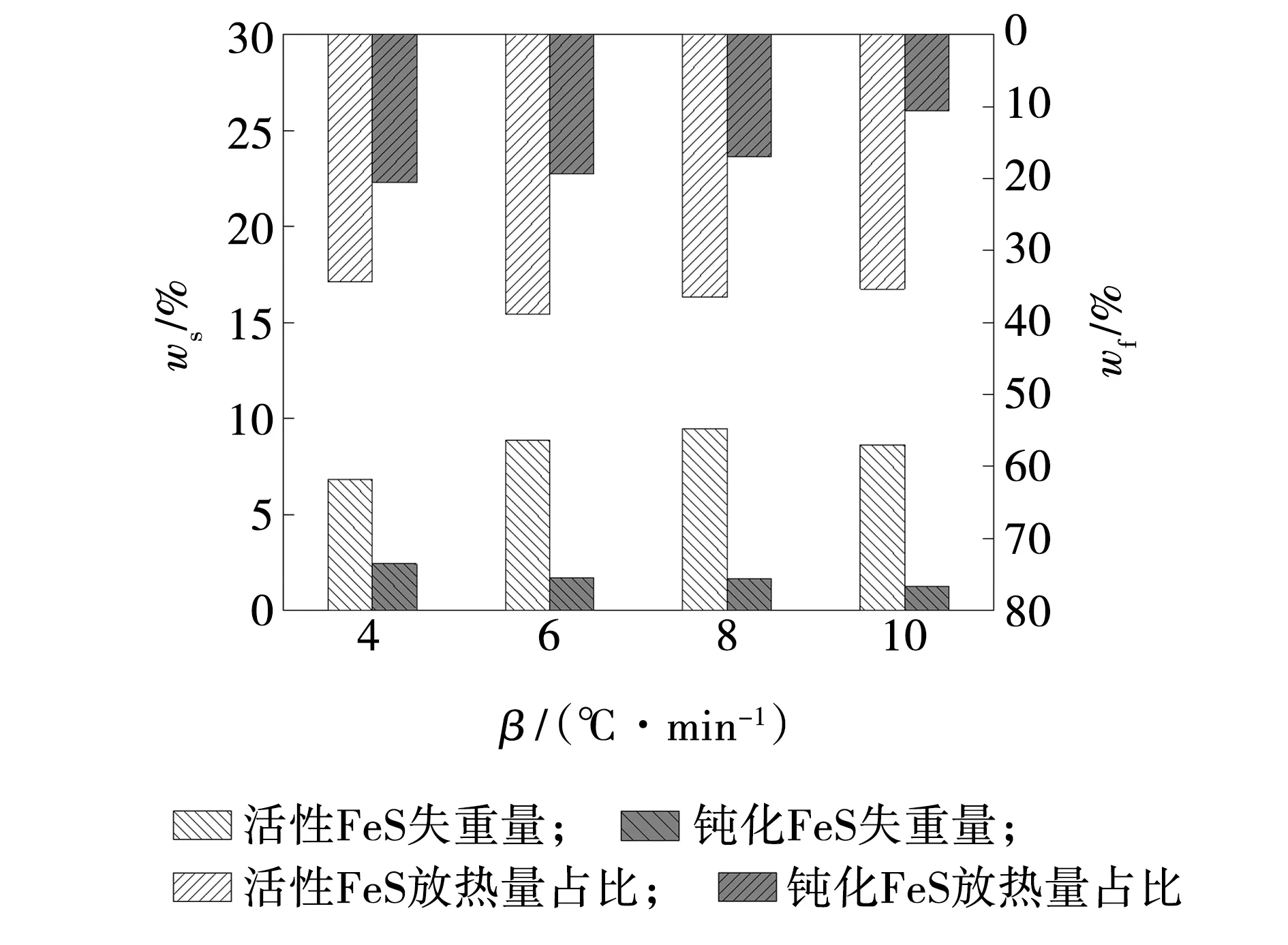
图10 FeS吸氧增重阶段增重量及放热量占比Fig. 10 Weight increment and heat released proportion of FeS at weight gain and oxygen adsorption stages
随着升温速率增加,活性FeS吸氧增重量增多,但在10 ℃/min升温速率下吸氧增重量减少,说明过高的升温速率促进活性FeS氧化反应消耗氧气,耗氧量增多,吸氧增重量减少。相较于活性FeS,钝化后的FeS吸氧增重量显著降低,随着升温速率增加,吸氧增重量也在缓慢减少。经气相钝化处理后的FeS,吸附氧气的能力减弱,吸氧增重量大幅减少。即使升温速率增加,促进其与氧气结合反应,促进效果小于钝化处理对其吸氧增重的抑制效果,整体呈现随升温速率增加,吸氧增重量缓慢减少的状态。钝化FeS在吸氧增重阶段的放热量占比明显小于活性FeS,在 10 ℃/min升温速率下,钝化FeS的放热量占比较活性FeS降低了24.7%,说明钝化处理后的FeS在吸氧增重阶段放热大幅度降低,吸附氧气与之结合的能力大幅减弱,自燃活性大幅降低,充分表明钝化处理技术显著抑制FeS自燃活性。
2.4 气相钝化对FeS分解燃烧阶段的影响
FeS吸氧增重达到最大值后进入了快速分解燃烧阶段,FeS与氧气反应加剧,生成了更多的二氧化硫气体,导致FeS质量减少,此阶段样品快速失重并快速释放热量,主要反应方程为

(1)
(2)
活性与钝化FeS分解燃烧阶段的持续温度及温度差值,如图11所示。
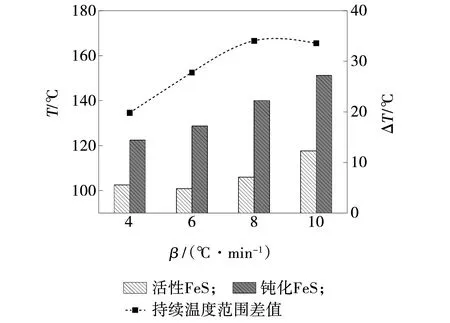
图11 FeS分解燃烧阶段的持续温度及温度差值Fig. 11 Sustained temperature of FeS at decomposition and combustion stages
活性FeS与钝化FeS在分解燃烧阶段的持续温度均随着升温速率增加而增大。钝化FeS的持续温度相较于活性FeS范围更广,在8 ℃/min升温速率下,钝化FeS持续温度较活性FeS延长34.06 ℃,说明升温速率越快,能够延长FeS分解燃烧持续的时间。两种样品的分解燃烧阶段的持续温度差值随升温速率增加呈增大趋势,在8 ℃/min升温速率下,达到温度差值最大值,10 ℃/min升温速率下,温度差值略微减少。钝化后FeS分解燃烧持续温度范围更广,说明气相钝化FeS生成的氧化膜存在于样品表面,分解燃烧由外向内依次进行,持续时间更长。
活性与钝化FeS分解燃烧阶段失重量及放热量占比,如图12所示。在分解燃烧阶段,活性与钝化FeS样品失重量随着升温速率增加总体呈减小趋势。活性FeS失重量在10 ℃/min升温速率下达到最低值,质量降低了15.8%。钝化FeS失重量保持在23%~25%范围,基本不随升温速率增加而产生明显变化。主要原因在于活性样品在室温下自燃生成铁氧化物,进入分解燃烧阶段的FeS占比相对较少,而钝化FeS因表面形成的氧化铁钝化膜,阻隔氧气,限制FeS室温下与氧气结合发生自燃,进入分解燃烧阶段的FeS相对较多,导致钝化FeS分解燃烧失重量增加。

图12 FeS分解燃烧阶段失重量及放热量占比Fig. 12 Mass loss and heat released proportion of FeS at decomposition and combustion stages
钝化FeS在其分解燃烧阶段放热量占比明显大于活性FeS,两种样品放热量占比随着升温速率增加都存在先减小后增加的趋势。在10 ℃/min升温速率下,活性FeS与钝化FeS放热量占比都达到最大值63.79%、87.70%。钝化FeS此阶段为整个自燃过程中主要放热阶段,放热占比最高达到87.7%,这说明气相钝化也能够导致FeS在分解燃烧阶段集中放热,导致其在分解燃烧阶段反应相对剧烈,放热量占比高。由于钝化处理后的样品表面存在钝化膜,在室温下基本不具备自燃的能力,缓慢吸附氧气致使FeS的自燃过程滞后,主要集中在180 ℃温度左右,因此在分解燃烧阶段,气相钝化处理后的FeS样品放热占比相较于活性样品更高。
2.5 气相钝化对活性FeS自燃过程活化能的影响
FeS自燃过程活化能的大小能够用来衡量其自燃倾向性[19],采用Kissinger法计算活性FeS与钝化FeS自燃过程的活化能。
Kissinger 法假设f(α)=(1-α)n,方程为
(3)
式中:f(α)——微分机理函数;
α——转化率;
E——活化能,kJ/mol;
β——升温速率,℃/min;
A——指前因子;
R——气体常数,8.314 5 J/(mol·K);
Tmax——升温速率β时对应的峰值温度,℃。
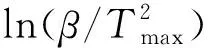

表1 不同升温速率下活性和钝化FeS的对应参数Table 1 Corresponding parameters of active and passivated FeS at different heating rates

图13 活性与钝化FeS线性拟合结果Fig. 13 Linear fitting results of active and passivated FeS
活性FeS的活化能46.17 kJ/mol,气相钝化FeS的活化能97.07 kJ/mol,指前因子分别为1.09×107、1.69×109。活化能大小可以衡量物质自燃倾向性,活化能越小越易自燃。经过气相钝化后的FeS自燃的活化能远大于活性FeS,说明气相钝化有效抑制FeS的自燃活性,从而降低其自燃危险性。
3 结 论
(1) 活性FeS的自燃过程分为自燃失重、吸氧增重、分解燃烧失重和燃尽四个阶段。活性FeS自燃过程有3个放热峰,钝化FeS存在吸氧增重阶段较小放热峰和分解燃烧较大的放热峰。气相钝化的FeS室温下无法自燃,自燃放热初始温度由室温后移至180 ℃,有效延长FeS自燃的诱导期,气相钝化能够有效抑制活性FeS的自燃过程。
(2) 活性FeS室温下自燃小幅失重并且迅速放热,随后进入吸氧增重阶段。气相钝化FeS生成的钝化膜阻隔FeS吸附氧气,钝化FeS吸氧增重量明显降低,吸氧增重阶段持续时间减少,氧化诱导期延长,吸氧增重阶段放热量大幅降低,气相钝化能够有效抑制FeS自燃过程前期结合氧气能力。
(3) FeS吸氧增重达到最大值后进入了快速分解燃烧阶段,大幅失重的同时释放大量热。气相钝化FeS分解燃烧阶段的持续时间、失重量高于活性FeS。气相钝化处理FeS生成的钝化膜附着于表面,缓慢吸附氧气后导致FeS自燃过程滞后,集中在180 ℃左右放热,放热明显高于活性样品。
(4) 活性FeS自燃过程活化能为46.17 kJ/mol,气相钝化FeS自燃过程的活化能97.07 kJ/mol,相较活性FeS大幅增加110.24%。气相钝化处理后的FeS活化能显著升高,氧化自燃更加稳定,自燃活性得到有效抑制,危险性大幅降低。
